Pharmacoresistant Epilepsy: A Current Update on Non-Conventional Pharmacological and Non-Pharmacological Interventions
Article information
Abstract
Uncontrolled seizure or epilepsy is intricately related with an increase risk of pharmacoresistant epilepsy. The failure to achieve seizure control with the first or second drug trial of an anticonvulsant medication given at the appropriate daily dosage is termed as pharmacoresistance, despite the fact that these drugs possess different modes of action. It is one of the devastating neurological disorders act as major culprit of mortality in developed as well as developing countries with towering prevalence. Indeed, the presence of several anti-epileptic drug including carbamazepine, phenytoin, valproate, gabapentin etc. But no promising therapeutic remedies available to manage pharmacoresistance in the present clinical scenario. Hence, utility of alternative strategies in management of resistance epilepsy is increased which further possible by continuing developing of promising therapeutic interventions to manage this insidious condition adequately. Strategies include add on therapy with adenosine, verapamil etc or ketogenic diet, vagus nerve stimulation, focal cooling or standard drugs in combinations have shown some promising results. In this review we will shed light on the current pharmacological and non pharmacological mediator with their potential pleiotropic action on pharmacoresistant epilepsy.
Introduction
Epilepsy is a common but fatal neurological disorder in humans after cerebral stroke. Epilepsy affects about more than 60 million people worldwide whereas more than 40% patients confirm pharmacoresistance during the course of their recurrently lifelong condition. In spite of the availability of different class of anti-epileptic therapies, pharmacoresistant epilepsy becomes a major medical trouble which is waiting for satisfactory management. Seizure is one of the important symptoms of epilepsy, characterised as uncontrolled electrical activity in the brain, which leads to either physiological convulsion, minor physiological signs, thought disturbances or combinations of symptoms.1–3 Numerous treatments including carbamazepine, valproate, phenytoin etc are available which can obstruct the neuroexcitability by preventing GABAergic system (Benzodiazepines, Barbiturates, Tiagabine, and Vigabatrin) or block the sodium channels Carbamazepine, Lamotrigine and Phenytoin).4 The condition in which seizure management is not achieved by first or second drug trial of anticonvulsant medication at daily therapeutic dosage, is termed as pharmacoresistance.5–7
Pharmacoresistance epilepsy is associated with five-fold higher mortality rate when compared to general population.8 The causes of drug resistant epilepsy are numerous, many due to abnormalities in brain maturation, severe brain injuries with resultant irreversible changes to cerebral neuroglia organization and inhibitory neuron function, kindling phenomenon, seizure-induced disturbances of oxygen supply, as well as acquired (or hereditary) changes in transporter proteins of blood-brain barrier (BBB) which function in the efflux of AEDs from the brain.9 Thus some pivotal strategies including resensitization of neurons towards drugs or molecular advancements are needed to treat this insidious condition.
Grounds of pharmacoresistance epilepsy
Several mechanisms have been proposed to explain pharmacoresistance like disease related mechanisms, drug related mechanisms and genetics. The transporter hypothesis relates to pharmacokinetic aspects, which suggests that over expressed efflux transporters at the BBB limits brain penetration of AED’s.10,11 One of the glycoprotein active efflux systems i.e. P-glycoprotein (P-gp) consists group of closely related intrinsic membrane proteins which are involved in multidrug resistance and encoded by a small family of multidrug resistance are encoded by MDR1 and mdr1a genes in human and rodent respectively.12 Preclinical study reveals that the absence of its substituent (mdr1a or mdr1b genes) shows a significant increase in-uptake of various lipophilic drugs in brain.13 Fig. 1A illustrate ATP dependent multi drug transporters such as P-gp or MRP2 located at the apical cell membrane of the endothelial cells acts as an active efflux pump by transferring part of drug back into the blood, thus limiting penetration of many lipophilic drugs into brain parenchyma. By lowering the concentration of drug in endothelial cells, multidrug transporter proteins also promotes flux from brain extracellular space into endothelial cells. Moreover, as Fig. 1B shows these transporters are over expressed and may contribute to barrier function. Hence, AED concentrations would be insufficient to cause antiepileptic potential.14–21

Representation of different mechanisms associated with resistance development in epilepsy (A) shows the normal activity of transporter protein (P-gp transporter) and sufficient concentration at target site; (B) shows the over-expression of transport protein leading reduced concentration of AEDs at target site; (C) represents the normal target protein orientation and hence good efficacy of AEDs (D) represents the modified orientation of target protein leading inefficacy of AEDs; (E) represents overall mechanisms for the development of pharmacoresistance in epilepsy.
On the other hand target hypothesis contends that target receptor sites are also altered in the epileptic brain which may cause insensitivity to anticonvulsant effects (as shown in Fig. 1C: normal, d: altered target).22,23 The main targets of the current AEDs are voltage gated ion channels and neurotransmitter receptors.24 After permeation into CNS parenchyma drugs have to bind to one or more target molecules to exert their desired action. But due to molecular changes in drug targets whether intrinsic (Polymorphisms) or acquired (by disease process, seizure, drugs etc.), may leads to reduced sensitivity.22 Thus, AEDs failed to block excitatory sodium or calcium currents or to enhance GABA mediated inhibition in pharmacoresistance patient.23
Another aspect related to pharmacoresistance pathogenesis is Hippocampal Sclerosis associated with Temporal lobe epilepsy (TLE). It is one of the most prevalent forms of epilepsy in this extensive neurodegenration in Cornu Ammonis areas: CA1 and CA3 areas of hippocampus and in the hilus of dentate gyrus take place. Clinical studies indicate that pharmacoresistance associated with TLE is because of failure of neurotransmission mediated by glutamate and GABA.25 Physiologically, the constituent subunits of GABAB receptors GABAB R1 (GBR1) and GABAB R2 (GBR2) are distributed across the dendritic layers of CA1 and CA3 and dentate gyrus. GABA binds to its receptor GABAB receptors and significantly controls the neuronal excitability. These signaling cascades mediate presynaptic regulation of neurotransmitter release as well as postsynaptic inhibition through G protein mediated activation of potassium channels and reduction of adenylyl cyclase activity. In-addition, increased level of glutamate in seizures can cause cell death, synaptic reorganization and further aggravated seizure activity.25,26 In summary, alterations in expression of MPR1, MPR2 and other genes associated with formation of different subunits of channels or receptors as shown in Fig. 1E could lead to pharmacoresistance.
Pharmacological intervention for pharmacoresistance
Verapamil
Verapamil, is a L-type calcium channel blocker of phenylalkylamine class which has identified as P-gp inhibitor and can increase intracellular concentration of AED’s (shown in Fig. 2A. Combined effect of verapamil and carbamazepine (CBZ) therapy has been shown greatly improved seizure symptoms in AED resistant patient.27 A case study reports that verapamil can vanished the electrical status epilepticus in an 11 year old boy who had resistance to AEDs.28 Recent finding with a pilot study in children suggest that add on therapy of verapamil seem to have regulatory effect in the drug resistance epilepsy.29 Thus, verapamil may offer a new hope for pharmacoresistant patients by improving seizure control due to its potential P-gp inhibitory effects.8,22
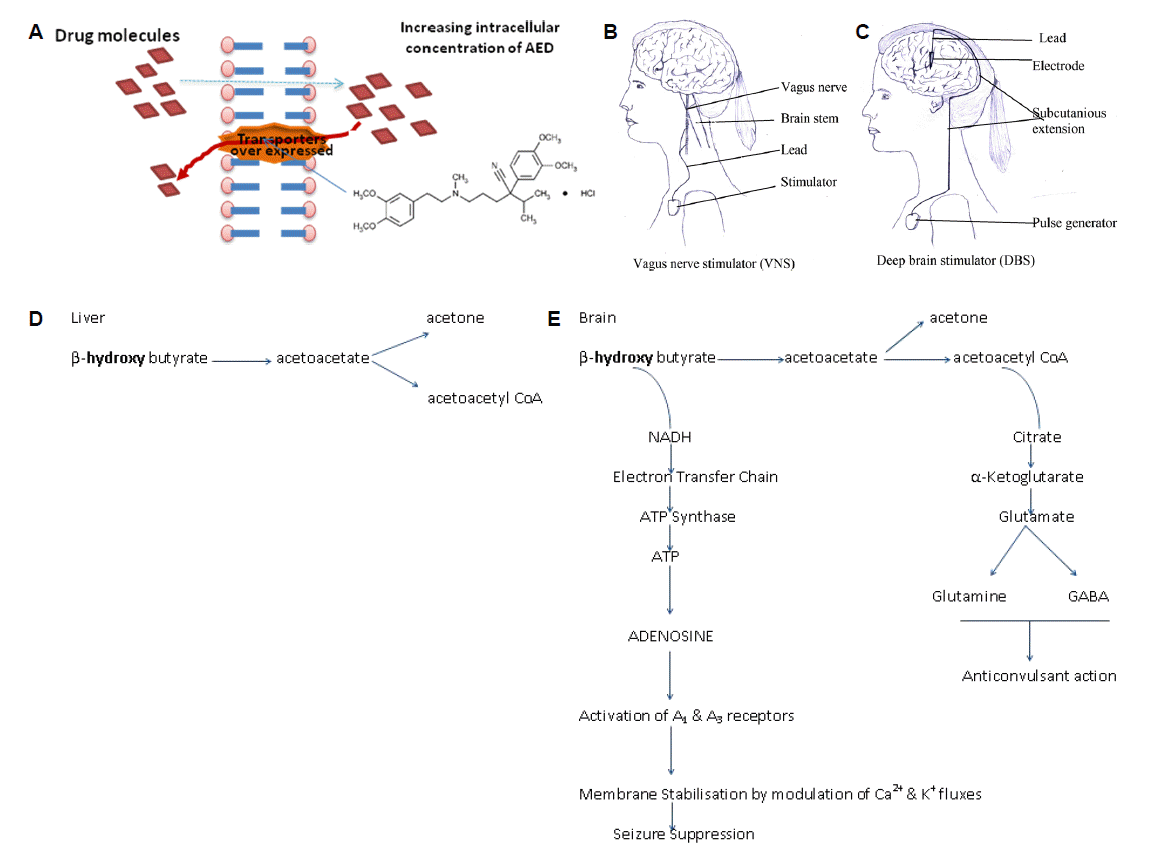
Representation of strategies to treat drug resistance in epilepsy (A) represents the inhibition of over expressed P-gp transporters; (B) represents electrical circuit for vagus nerve stimulation and (C) deep brain stimulation; (D) represent normal processing of ketone bodies in liver and (E) in brain where these metabolites further converted into product (adenosine and GABA) leading to reduction in epilepsy.
Atorvastatin
Atorvastatin, aninhibitors of 3-hydroxy-3-methyl-glutaryl-CoA re-ductaseuse to cholesterol level in blood. In-addition it has been found that atorvastatin also inhibits N-Methyl-D-aspartate (NMDA) mediated excitotoxicity in vitro neuron culture. The pileotropic neuroprotective action of statins exert their effects by inhibiting synthesis of isoprenoids which further interfering with isoprenylation and membrane attachment of small guanosine triphosphate (GTPases). Although it has well known that atorvastatin can significantly decline the threshold of seizures when administered against pentylenetetrazol30–32 but another study also revealed that atorvastatin majorly inhibits hippocampal cell death induced by kainic acid (an agonist for a subtype of inotropic glutamate receptor, AMPA and kainic acid receptor) in CA1 and CA3 regions and produce neuroprotection, anti-excitotoxicity and antiepileptic action.33
NeuropeptideY
NeuropeptideY (NPY), is a 36 amino acid peptide which is widely distributed in the nervous system and plays an important role of a neurotransmitter and neuromodulator.34 It is mainly produced and released by GABAergic interneurons and inhibits glutamatergic neurotransmission in the excitatory trisynaptic circuit. In epileptic conditions there is robust overexpression of NPY receptors in the granular and pyramidal cells consequently lead to tonic inhibition of glutamate release.35–37 Several recent research finding reveals NPY receptor antagonists may be used for treating pharmacoresistant forms of TLE.38,39
Adenosine
Adenosine plays an important role in biochemical processes such as energy transfer. It is a brain endogenous anticonvulsant that is considered to be responsible for seizures arrest and postictal refractoriness. Adenosine and its analogues can suppress pharmacoresistant seizures but most of these pharmacological approaches are limited to strong systemic side effects so activation of central adenosine A1 receptors leads to the suppression of seizure activity in mouse model of drug resistant epilepsy.40
Thyrotropin-releasing hormone (TRH) and its analogs
TRH (L-pyroglutamy- L-histidyl-L-prolinamide), a tripeptide, is the first hypothalamic releasing hormone. It is a tripeptide having a dual role as a hormone and as a neuropeptide. Role of TRH in epilepsy is marked by increase prepro-TRH messenger ribonucleic acid (mRNA) and peptide level in animal model of epilepsy. TRH mediates its functions by binding to receptors TRH-R1 and TRH-R2, which are the members of rhodopsin/b-adrengeric receptor subfamily. TRH and some of its analogs have been reported to protect against PTZ, 4-amino pyridine (4-AP), kainic acid and glutamate-induced seizures. 24,41–43 In addition, TRH also play a significant role in management of different type of intractable epilepsy or epileptic syndromes including infantile spasms (unique, and frequently malignant, epileptic syndrome confined to infants.), Lennox- Gastaut syndrome (form of childhood-onset epilepsy that most often appears between the second and sixth year of life, characterized by frequent seizures, myoclonic seizures, generalized seizures and refractory partial seizures.44 Furthermore, Deng et al. (2007) has revealed that TRH may increase the GABA release in CA1 regions and inhibit resting membrane K+ conductance which leads to excitability of interneurons and facilitated GABA release.45
Non pharmacological interventions for pharmacoresistance
Surgical procedures
Surgery may be the most apposite treatment option in some cases especially where strong evidence exists for its efficacy but several factors need to be taken into account alongside the potential benefits.46 Major surgery including corpus colostomy may be considered as last significant treatment option in palliative care for intractable seizures. A multicenter study of 339 epileptic patients for 2 years reported that 68% of the patients who received temporal lobe lobectomies experienced seizure remission, compared to 50% of the patients who underwent extra temporal resections. Surgical procedures are generally considered after the failure of two appropriate drug regimens. Risk and safety parameter should be measurement in each case study for more primitive data to guide therapeutic decision.47
Neurostimulation
Neurostimulation including vagus nerve stimulation (VNS) and Deep brain stimulator (DBS) are the non pharmacological adjunctive approaches for the treatment of pharmacoresistance patients as shown in Fig. 2B and 2C respectively. This is a reserved therapy for patients who cannot undergo in surgery and for whom, surgery has failed.46,48 Neurostimulation is carried out by short bursts of electrical energy directed into the brain via vagus nerve. Neurocybernetic prosthesis (NCP), a vagal nerve stimulator is implanted in the patient’s chest on the upper left side as shown in Fig. 2). Preclinical study reported that the stimulation of vagal nerve fibers (about 4Hz frequencies) can prevent experimental induced clonic seizures by intra-peritoneal injection of PTZ and tonic-clonic seizures caused by maximal electroshock (MES). This preventive effect of vagus nerve stimulation may be act through transient desynchronization of cortical rhythms or by raising the seizure threshold by releasing glycine and GABA in the brain structures.47
Deep brain stimulation involves electrical stimulation of specific subcortical nuclei, which have widespread neural connections. In a randomized controlled trial of 109 patients with refractory epilepsy who had DBS electrodes implanted in the anterior nucleus of the thalamus there was 29% greater reduction in seizures for 54 patients, who had the stimulator switched as compared to 55 patients who had their stimulation turned off after a blinded period of 3 months.47,49
Unlike other methods of neurostimulation, responsive neurostimulation (RNS) does not deliver electrical stimulation at specific frequencies throughout the day, instead the RNS device is composed of a combined recorder and stimulator device which detects clinically relevant epileptiform discharges and delivers appropriate electrical stimuli in response. This non-invasive neurostimulation technique with low frequency repetitive transcranial magnetic stimulation (rTMS) might suppress cortical excitability either via inducing long term depression or by enhancing GABAergic inhibition, but the evidence for its efficacy is still limited.47
Focal cooling
By focal cooling of the brain reduced epileptiform activity in animals and may also terminate focal seizures.50,51 It has been observed that rapid cooling to 22°C can stop seizure activity induced by 4-AP. In-addition Javedan et al. reveal that focal cooling with thermoelectric chips (Meclor, Trenton, NJ, USA) each 3.5×3.5×2.4 mm in dimension and was attached together to the end of copper rod may abolish focal seizures.51 The site of the 4-AP injection was either cooled intermittently (Post Cool) for 30 s every 2 min, starting 4 min after the 4-AP injection or precooled (Precool) for 30 min before 4-AP injection, with same frequency of 30 sec cooling cycle. These experiments results defend that cooling the brain can rapidly terminate focal seizures.52
Dietary therapies
There are four types of metabolic therapies used for the reduction of seizures including These are classic ketogenic diet, the medium chain triglyceride (MCT) diet, the modified Atkins diet (MAD) and the low glycemic index treatment (LGIT).47 The ketogenic diet has been used as a therapeutic alternative to antiepileptic drugs (AEDs) for the treatment of refractory epilepsy and usually reserved for young patients with difficult to control seizures. The ketogenic diet is a special high fat, low carbohydrate diet typically in a ratio of 4:1 or 3:1. It gets converted to ketone bodies (beta -hydroxy butyrate or aceto acetate) and also increases GABA activity in the brain.53 In MCT more carbohydrates and proteins can be consumed for every gram of fat and there is no limit on the amount of protein consumed or the total number of calories consumed per day. MCT diet is more successful in the treatment of children patient due to its acceptability as compared to adults. The MAD diet has a fraction of 1 gram of fat for every gram of carbohydrates plus protein and is less restrictive than other two fat diets. The low glycemic index treatment (LGIT) restricts carbohydrate intake to food items with a glycemic index of less than 50 at 40–60 grams per day.47,54–56 Dietary therapies show some side effects like constipation, acidosis, kidney stones, temporary hypercholesterolemia and hunger. But in-addition to showing high levels of efficacy similar to many AEDs, the dietary interventions are generally quite tolerable and demonstrate the effectiveness in a relatively short time period.47 The possible mechanisms of protection by dietary therapies is because of difference in processing of ketonebodies in brain as shown in Fig. 2E than in liver shown in Fig. 2D.
Herbal remedies
Herbal remedies are another divine source for the management of various insidious diseases including pharmacoresistance epilepsy. Various substances such as herbs, foods, vitamins, energy medicines, animal compounds act as complementary and alternative therapy for epilepsy. In a randomized double blind controlled trials cannabinoids have shown supporting evidence for their anticonvulsants action.57 Moreover, huperzine A, kava (Piper methysticum) and mistletoe (viscumsp) shows good laboratory results.47,57,58 As India being a rich source of fauna and flora, so could be a great source of future medicines not only for resistant epilepsy but also for other untreatable diseases. Thus a window of research is open at this gateway for the potential substance against pharmacoresistance epilepsy.
Conclusions
Despite the enormous efforts had been made and continuing by major pharmacy industry as well as reported academic institution, a huge number of individuals still falls in category of refractory epilepsy. Currently there is no drug which can prevent epileptogenesis so early identification of etiology and form of epilepsy would be helpful in appropriate treatment. Some of these patients could benefit from non-pharmacological treatment modalities, especially epilepsy surgery. Indeed, resective surgery in refractory temporal lobe epilepsy has been shown in a randomized study which provides substantially better outcomes than continued manipulation of AEDs therapy. When pharmacological and surgical approaches are not feasible, stem cell therapies might be an alternative and also offers the additional potential for cell replacement and neural circuit repair. For the treatment of hippocampal sclerosis, human embryonic stem cells (hESCs) offer hope that a stem cell cure may be on the horizon for treating some forms of resistant epilepsy.
Acknowledgements
We express our gratitude to Honorable Founder President Dr. Ashok K. Chauhan, RBEF Chairman, AKC Group of Companies, Amity University, Noida, India, for their inspiration and constant support for this study.
Notes
Conflict of Interest Statement
No conflict of interest has been declared.