Introduction
Epilepsy is one of the most common neurological disorders affecting 1–2% of the world population, and almost two million people in the United States alone.1 Excluding the small percentage of people who underwent successful resection of epilepsy surgery, the majority of patients are treated with various antiepileptic drugs (AEDs), which impose safety concerns over the duration of treatment. Despite the advent of new AEDs over the past 20 years, still more than 30% of epilepsy patients experience recurrent seizures2,3 and many experience undesirable side effects. Therefore, there are still unmet needs for the treatment of epilepsy and there remains a need to develop new treatment methods that could reduce seizure frequency with improve safety profile. For those patients with medically refractory epilepsy, combined administration of AEDs and the use of new AEDs are the most appropriate therapeutic options. There are several new AEDs introduced last several years and there are more than dozen of new AEDs under development, which include brivaracetam, carabersat, carisbamate, eslicarbazepine acetate, ganaxolon, huperzine, lacosamide, losigamone, remacemide hydrochloride, ezogabine (retigabine), perampanel, rufinamide, safinamide, seletracetam, soretolide, stiripentol, talampanel, tonabersat, and valrocemide. In addition, new devices are now available that stimulate certain parts of the brain directly. The article reviews their pharmacokinetic profiles, drug interactions, molecular mechanisms of action, efficacy, and tolerability.
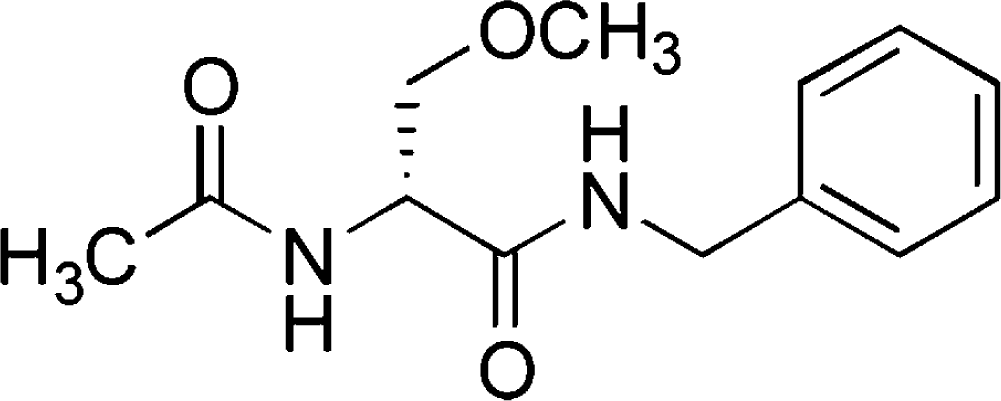
Lacosamide
Lacosamide (LCM) is a new antiepileptic drug that was approved as an adjunctive treatment for partial-onset seizures for patients ≥16 years by the European Medicines Agency (EMEA) in August 2008 and for patients ≥17 years by the U.S. Food and Drug Administration (FDA) in October 2008. It is now approved in more than 25 countries in the world and available in oral tablet, IV solution, and oral syrup. LCM has a novel mode of action (MOA) that appears to be different from existing AEDs.
Pharmacokinetics
LCM has a linear pharmacokinetic profile with high oral bioavailability.4 Studies in healthy volunteers demonstrated that LCM is almost completely absorbed, and the absorption is not affected by the presence of food.5 Peak serum concentrations occur at 1–2 hours after oral intake, and the elimination half-life of LCM is about 13 hours, allowing convenient twice-daily dosing.6 LCM intravenous solution is typically administered over 30 minutes and the Cmax is reached at the end of infusion. LCM has low plasma protein binding (≤15%) and the volume of distribution is approximately 0.6 L/kg, which is similar to body water.7 Following twice-daily administration of oral LCM, steady-state plasma concentrations are reached after 3 days.
Lacosamide is primarily eliminated renally as unchanged drug (>40%) and an inactive metabolite, O-desmethyl metabolite (<30%).4,6 Although the hepatic isoenzyme 2C19 is mainly responsible for the formation of the O-desmethyl metabolite, co-administeration of CYP2C19 inducers or inhibitors did not cause clinically relevant differences in the pharmacokinetics of LCM, indicating that the metabolic pathway involving CYP2C19 is minor.
Mechanisms of action
Unlike other classical AEDs such as carbamazepine, phenytoin, and lamotrigine which act on fast inactivation of voltage-dependent sodium channels, LCM selectively enhances slow inactivation of voltage-dependent sodium channels without affecting fast inactivation.10
LCM demonstrated potent anticonvulsant activity in broad range of animal models of partial onset and pharmacoresistant seizures, generalized tonic-clonic seizures, as well as status epilepticus. Intraperitoneal LCM was effective in preventing seizures in the 6-Hz psychomotor seizure model (ED50 9.99 mg/kg) and audiogenic seizure model (ED50 0.63 mg/kg). LCM of 20 and 50 mg/kg completely prevented tonic convulsions induced by maximal electroconvulsive shock (MES) and 50 mg/kg provided partial protection against clonic convulsions induced by NMDA in mice.10,11 LCM was also effective in amygdala and hippocampal kindling models.11,12 However, LCM was less effective against clonic seizures induced by pentylenetetrazole (EC50-25 mg/kg), bicuculline (EC50>50 mg/kg), or picrotoxin (EC50>30 mg/kg) in rodents.10,11 LCM was also effective in self-sustaining status epilepticus model in rats, stopping seizures within 15 minutes of administration and preventing recurrence over 24 hours.10
Efficacy and tolerability
Three pivotal studies (one phase II and two phase III studies) have been conducted to establish the efficacy and safety of LCM.13–15 Three doses of LCM (200, 400, and 600 mg/day) were administered as adjunctive therapy for patients with partial epilepsy with or without secondary generalization, with a starting dosage of 50 mg BID, followed by weekly increase of 100 mg/day to the target dose. Total of 1,294 patients were randomized in these studies and up to three concomitant AEDs were allowed.
The primary efficacy analysis was conducted on the intent-to-treat (ITT) population, which is defined as all randomized patients who received at least one dose of the trial medication and had at least one post-baseline efficacy assessment. In the phase II study, the 50% responder rates were 32.7% for 200 mg/d (p=0.090), 41.1% for 400 mg/d (p=0.004), and 38.1% for 600 mg/day (p=0.014), compared with 21.9% for the placebo group.13 Percent reduction in seizure frequency per 28 days over placebo was 14.6% in the LCM 200 mg/day group (p=0.101), 28.4% in the LCM 400 mg/day group (p=0.002) and 21.3% in the LCM 600 mg/day group (p=0.008) groups. Two subsequent phase III studies confirmed the efficacy and safety of LCM at doses of 200–600 mg/day.14,15 Subsequent analysis of pooled efficacy data from these trials further supports the overall efficacy of LCM at doses of 200–600 mg/day.16 For the pooled analysis, the 50% responder rates were 22.6% for placebo, 34.1% for LCM 200 mg/day, and 39.7% for LCM 400 mg/day. The median percent reduction in seizure frequency was 18.4% for placebo, 33.3% for LCM 200 mg/day, and 36.8% for LCM 400 mg/day.16 Overall, the LCM 600 mg/day group showed similar efficacy to the 400 mg/day group. Pooled analysis demonstrate that complete seizure freedom during the maintenance period was achieved in 2.7%, 3.3% and 4.8% of patients randomized to LCM 200, 400, and 600 mg/day, respectively, compared with 0.9% in the placebo group.16
The most common treatment emergent adverse effects (TEAEs) of oral LCM were dizziness, headache, nausea and diplopia. All of these TEAEs were dose-related except for headache, and reported more often during titration rather than during the maintenance phase. The incidence of somnolence during the treatment period was approximately 7% for placebo and 9% for the total LCM groups, and did not appear to be dose-related. The incidence of rash was low for patients randomized to LCM (3%), and was similar to that reported with placebo. No rashes were serious and all were assessed as mild to moderate in intensity. LCM appears to have no significant cardiac side effects. Evaluation of ECG readings demonstrated little change from baseline to the end of maintenance in heart rate, QTc interval or QRS duration for the placebo and LCM groups. A small increase in mean PR interval at the end of maintenance (1.4–6.6 ms increase) was noted. There were no reports of adverse events associated with PR interval prolongation, and the degree of increase is considered to be similar to other AEDs.17
TEAEs associated with intravenous LCM were mild or moderate in intensity and included dizziness, headache, back pain and somnolence. Infusion site-related pain was infrequent (0% in 60 min and 11% in 30 min infusion), and did not result in discontinuations of LCM.18 In another open-label study (n=60), in which LCM was infused faster over 10, 15, or 30 minutes for 2–5 days (200–800 mg/day), the incidence of adverse events was similar with headache (5%, 7%, 8%) and dizziness (5%, 6%, 8%) being most commonly reported.19
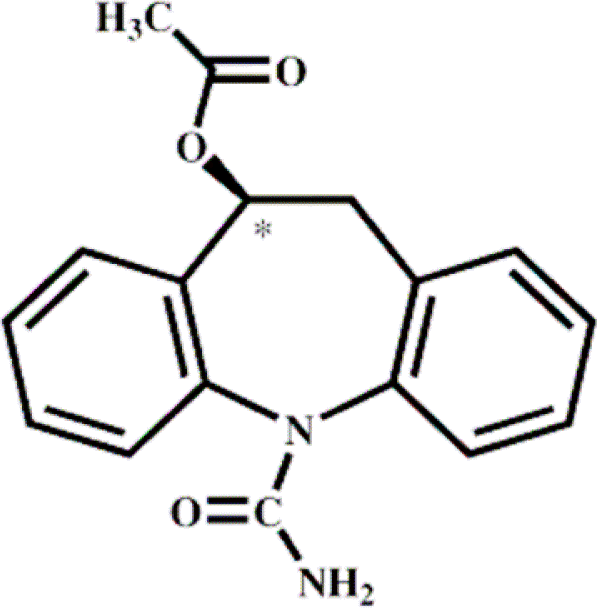
Eslicarbazepine acetate
Eslicarbazepine Acetate (ESL) is a prodrug of eslicarbazepine, which shares structural similarity with carbamazepine and oxcarbazepine (Table 1). Therefore, ESL is considered as a third-generation, single enantiomer member of the established family of dibenz/b, f/azepine AEDs represented by carbamazepine and oxcarbazepine.20 ESL was licensed in April 2009 by the EMEA as an adjunctive therapy for partial seizures in patients ≥18 years, but has not yet been approved by the FDA.
Pharmacokinetics
ESL is rapidly and extensively metabolized to eslicarbazepine by hydrolytic first-pass metabolism within 1–4 hours.21 Unlike carbamazepine, ESL is not metabolized to carbamazepine-10, 11-epoxide and is not susceptible to metabolic autoinduction.22 Unlike oxcarbazepine, which is a prodrug to both eslicarbazepine (also called S-licarbazepine or S-MHD) and R-licarbazepine (also called R-MHD), ESL is a prodrug of eslicarbazepine, which is responsible for pharmacological activity. The pharmacokinetics of eslicarbazepine is linear and almost completely absorbed (>90%) with or without food.23 The volume of distribution of eslicarbazepine is about 34 liters and protein binding is estimated to be less than 40%.21 In adult epilepsy patients, the half-life of eslicarbazepine was 13–20 hours and the steady-state concentration was reached within 4–5 days of once daily dosing.21 The metabolites of ESL are primarily excreted through kidney in unchanged form and glucuronide conjugates (30%). As their clearance is dependent on renal function, dosage adjustment may be necessary in patients with creatinine clearance below 60 mL/min.24
ESL does not inhibit the activity of hepatic isoenzymes but a moderate inhibitory effect was seen on CYP2C19.25 ESL may also have a mild inducing effect on CYP2C9 since coadministration of ESL with warfarin has been demonstrated to decrease serum level of (S)-warfarin.25 Pharmacokinetics analysis from phase III studies showed no relevant effect of ESL on the clearance of carbamazepine, phenytoin, topiramate, clobazam, gabapentin, phenobarbital, levetiracetam nor valproic acid.26 In addition, protein binding of eslicarbazepine was not affected significantly by the presence of warfarin, diazepam, digoxin, phenytoin and tolbutamide.25 However, plasma concentrations of oral contraceptives (both ethinylestradiol and levonorgestrel) are reduced after daily dose of ESL 1,200 mg, with the AUC decreased by 32 and 24%, respectively.21
Mechanisms of action
Although the precise MOA of ESL is not known, in vitro studies suggest that ESL interacts with the inactivated state of a voltage-gated sodium channels.27 Previous studies in rat striatal slices demonstrate that ESL acts similarly to carbamazepine and oxcarbazepine by inhibiting the release of some neurotransmitters or neuromodulators, such as glutamate, GABA, aspartate and dopamine.28
ESL demonstrates anticonvulsant activity in several animal models. It blocks tonic seizures in the MES model and limbic seizures in the corneal kindled mouse and amygdala-kindled rat.21 However, ESL displays only weak effects against clonic seizures induced by PTZ, bicuculline, picrotoxin, and 4-aminopyridine.25
Efficacy and tolerability
A phase II placebo-controlled study found that ESL could be an efficacious and well-tolerated treatment option for patients with refractory partial-onset seizures.29 In this study, the percentage of responders showed a statistically significant difference between ESL and placebo groups (54% vs. 28%, p=0.008). In the subsequent phase III trial, patients were randomized to placebo (n=102) or once-daily ESL 400 mg (n=100), 800 mg (n=98), or 1,200 mg (n=102) in the double-blind treatment phase.30 The starting dose of ESL was 400 mg with weekly increase of 400 mg to the full target doses. Responder rates were 20% (placebo), 23% (400 mg), 34% (800 mg), and 43% (1,200 mg). Median reduction in seizure frequency was 16% (placebo), 26% (400 mg), 36% (800 mg, p= 0.359), and 45% (1,200 mg, p=0.0009). The most frequent concomitant AEDs were carbamazepine (56–62% of patients), and similar efficacy was obtained in patients administered ESL with or without carbamazepine. Two other phase III trials in 23 countries also showed similar results. Combined analysis demonstrated that ESL dosages of 800 and 1,200 mg once daily significantly reduced median seizure frequency reduction compared to placebo (placebo: 8.5%; 800 mg: 29.4%, p<0.0001; 1,200 mg: 30.6%, p<0.0001). However, no significant difference in responder rate was found between the ESL 400 mg and placebo arms in any study.25
The most commonly reported TEAEs were dizziness (18.8%), somnolence (11.2%), nausea (6.5%), diplopia (6.3%), headache (5.5%), vomiting (4.8%) abnormal coordination (4.4%), blurred vision (3.5%), vertigo (2.1%) and fatigue (2.1%).29–31 Overall, TEAEs of ESL were mild to moderate and appeared to be dose dependent.49,50 The incidence of psychiatric complications, rash, or hyponatremia was low (<1% of patients).25,31 No difference in tolerability has been reported between adults and elderly patients, and no abnormal vital signs were seen in patients on ESL.21,31 There were no significant changes in the laboratory parameters or ECG parameters.
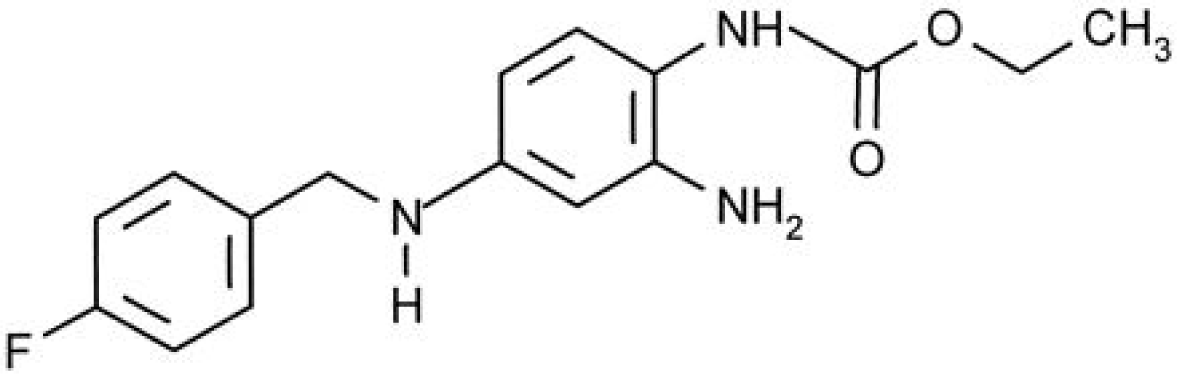
Ezogabine (retigabine)
Ezogabine (EZG) is a new antiepileptic medication with a novel mechanism of action. It is also known as retigabine and previously as a chemical compound D-23129, which was developed as adjunctive treatment for partial epilepsy. EZG’s anticonvulsant properties are primarily mediated by augmenting neuronal voltage-gated potassium channels. EZG was recently approved as an adjunctive treatment for partial-onset seizures for patients by the EMEA in March 2011 and by the FDA in June 2011.
Pharmacokinetics
EZG demonstrates a linear pharmacokinetic profile with dosage up to 1,200 mg/day.32 Although EZG is rapidly absorbed following oral administration, bioavailability of oral EZG is estimated to be only about 60%.32 Peak plasma concentration of EZG is delayed approximately to 2 hours with food, and modestly increased when EZG is taken with a high-fat meal.32 Protein binding is approximately 80% and the volume of distribution at steady-state is about 2–3 L/kg.32
EZG is metabolized by N-acetylation to the mono-acetylated metabolite (primary metabolite) and by glucuronidation to form N-glucronide structure, which demonstrates minimal pharmacologic activity.33 Both EZG and its primary metabolite have a plasma half-life of 8 hours (7.2 to 9.4 hours). The majority of drug and metabolites are excreted through kidney. EZG modestly inhibits CYP2A6 isoform.25 Renal clearance of EZG was reduced approximately 25% in individuals with mild renal dysfunction and approximately 50% in those with moderate or severe renal disease or those who required dialysis.25 In patients with moderate or severe hepatic impairment, EZG clearance is reduced by 30% to 50%.25
EZG does not induce or inhibit its own metabolism. Although EZG revealed no clinically significant pharmacokinetic interactions between EZG and valproate or topiramate, pervious study indicated that phenytoin and carbamazepine may increase the clearance of EZG by approximately 30% especially when higher dose of EZG (1,200 mg/day) is administered.34 In contrast, population pharmacokinetic analyses of data from all clinical studies involving more than 800 patients failed to identify any clinically meaningful effects of phenytoin and carbamazepine on EZG pharmacokinetics.35 In another study with healthy volunteers, lamotrigine mildly increased the half-life of EZG, while EZG increased lamotrigine clearance by about 20%. These findings are probably due to the fact that both medications are partially metabolized by glucronidation.36
Mechanisms of action
The anticonvulsant effect of EZG is primarily due to opening neuronal voltage-gated potassium channels, which enhances inhibitory M-type potassium current.37 EZG selectively enhances M-currents through heteromeric KCNQ2/3 channels and KCNQ3/5 channels38 as well as homomeric KCNQ5 channels.39 Potasium channel selectivity of EZG is important for the safety since KCNQ1 subunits are present in cardiac cells,40,41 while KCNQ4 subunits are present in the auditory system.42 On the other hand, mutations of KCNQ2/3 can results in an epilepsy syndrome known as benign familial neonatal convulsions.43
Although clinical trials were limited to partial onset seizures, in preclinical studies, EZG was effective against both partial seizure models (amygdala, corneal and hippocampal kindling models) and generalized seizure models (MES, pentylentetrazole, picotoxin, genetic epilepsy models), and a status epilepticus model (cobalt-homocysteine thiolactone model).44–46
Efficacy and tolerability
A double-blind, placebo-controlled, randomized phase II clinical trial evaluated three doses of EZG (600, 900, and 1,200 mg/day) administered as adjunctive therapy in adult patients with partial epilepsy with or without secondary generalization.47 In this study, the median percent change in seizure frequency of the ITT population was 23.4% for 600 mg/d, 29.3% for 900 mg/d (p=0.0387), and 35.2% for 1,200 mg/day (p=0.0024), compared with 13.1% for the placebo group.47 The difference was significant for EZG 900 and 1,200 mg/day arms when compared to placebo, but no significant difference was noted between the RTG 600 mg/day and placebo groups. Two additional phase III studies confirmed the dose-dependent efficacy of EZG (RESTORE 1 and RESTORE 2). In RESTORE 1 (n=301), median seizure frequency over placebo was significantly reduced in the ITT analysis: 44% for 1,200 mg/day (n=151) vs. 18% for placebo group (n=150),48 and in RESTORE 2, both 600 mg/day and 900mg/day doses showed a significant seizure reduction compared to placebo (p<0.001).49
In phase II study, most frequent TEAEs included somnolence (6% for placebo, 17% for 600 mg/day, 21% for 900 mg/day), headaches (9% for placebo, 14% for 600 mg/day, 19% for 900 mg/day dizziness (4% for placebo, 8% for 600 mg/day, 18% for 900 mg/day) confusion (5% for placebo, 5% for 600 mg/day, 8% for 900 mg/day), and asthenia (9% for placebo, 14% for 600 mg/day, 19% for 900 mg/day), followed by less frequent TEAEs including speech disorder, vertigo, tremor, amnesia, and abnormal gait.47 About 20% patients withdrew from the study due to TEAEs (mostly during the titration phase), and most common reasons for withdrawal included confusion, speech disorder, dizziness, and somnolence.47
Among non-CNS events, bladder-related adverse events (e.g. urinary hesitancy) were observed with EZG, primarily with the 1,200 mg arm. Bladder ultrasound revealed a modest increase in mean post-void residual volume at the 1,200 mg dose but not at lower doses. These may have reflected inhibition of bladder contractility and urinary retention secondary to EZG’s effects on KCNQ channels in the detrusor muscle of the bladder.25 Otherwise, there were no clinically relevant findings in laboratory measurements including urinalysis, ECG findings, or ophthalmologic examinations.
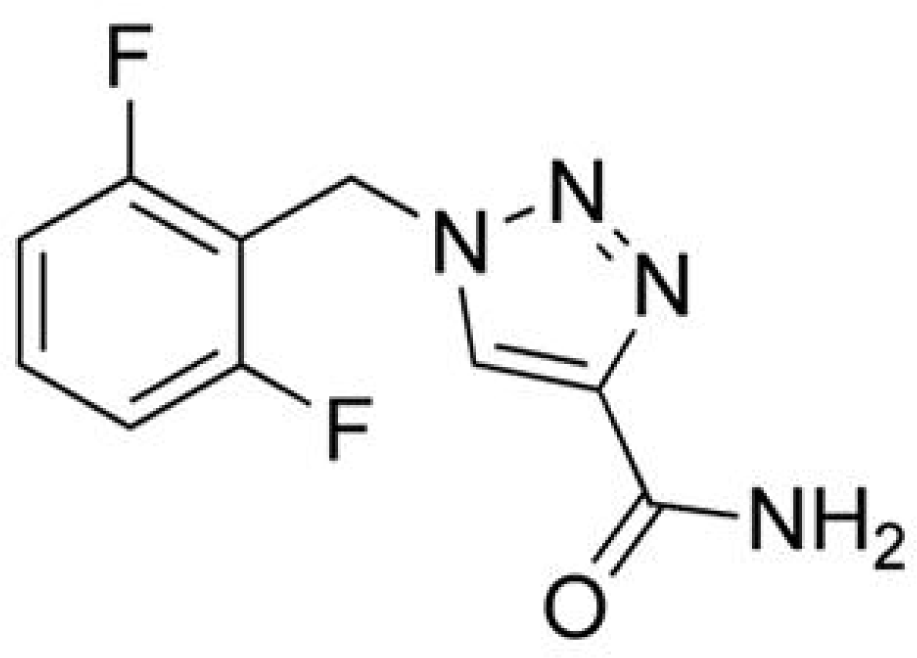
Rufinamide
Rufinamide (RFM; 1-[(2,6-difluorophenyl)methyl]-1H-1,2,3-triazole-4 carboxamide) is a novel compound that is structurally unique. RFM was granted Orphan Drug status in 2004 by both the EMEA and the FDA for the treatment of seizures associated with Lennox-Gastaut syndrome (LGS) in patients ≥4 years.50
Pharmacokinetics
RFM has a relatively slow rate of absorption and the maximum plasma concentration is reached in about 4–6 hours following oral administration under fasting or fed conditions. Gastrointestinal absorption of rufinamide is increased with food, and estimated absorption is about 85% under fed conditions.51 RFM has a relatively short half-life (6–10 hours) and plasma protein binding ranges from 26–35%. At a dose of 3,200 mg/day, the apparent volume of distribution for a 67kg subject with a body surface area of 1.79 m2 is estimated to be 52.7L.52 RFM is extensively metabolized through the hydrolysis of its carboxamide group by carboxylesterases into an inactive derivative that is mainly eliminated via renal excretion. Since its main metabolites are inactive, the pharmacokinetics of RFM are not affected by impaired renal function.52
RFM does not affect the plasma concentrations of topiramate or valproic acid, but may increase the oral clearance of carbamazepine (by 15% in children, 9% in adolescents, and 8% in adults) and lamotrigine (by 16% in children, 11% in adolescents, and 8% in adults) and may decrease the oral clearance of phenobarbital (by 12% in children, 9% in adolescents, and 7% in adults) and phenytoin (by 17.5% in children, 9.1% in adolescents, and 6.5% in adults). Furthermore, RFM decreases the plasma concentrations of norethindrone and ethyinyl estradiol. On the other hand, plasma concentration of RFM is decreased by carbamazepine (−23.2% in male children, −25.6% in female children, −18.6% in male adolescents, −20.4% in female adolescents, −20.5% in male adults, −21.8% in female adults) and vigabatrin (−26.8% in male children, −29.9% in female children, −17.1% in male adolescents, −18.4% in female adolescents, −13.7 in male adults, and −15.4% in female adults). Phenobarbital, phenytoin, and primidone also decrease the steady-state plasma concentration of RFM by 25–46%. In contrast, plasma concentration of RFM is increased by co-administration of valproic acid, especially in children.52
Mechanisms pf action
Rufinamide is structurally unrelated to other currently available AEDs; its exact mechanism of action in unknown. However, it is likely that rufinamide mediates its anticonvulsant effects by blockage of voltage-gated sodium channels and limiting neuronal sodium-dependent, high frequency action potential firing.53
Efficacy and tolerability
A pivotal, multicenter, randomized, double-blind, placebo-controlled clinical trial studied the efficacy of RFM as adjunctive treatment for seizures associated with LGS (n=139).54 The study consisted of a 28-day baseline period, followed by a 14-day titration period, where RFM was started at 10 mg/kg per day and titrated up to a maximum dosage of 45 mg/kg per day (maximum total daily dosage of 3,200mg), and a 70-day maintenance period. The most frequently used concomitant AEDs were valproate, lamotrigine, and topiramate. This clinical trial demonstrated that RFM significantly reduced the median total seizure frequency (32.7% vs. 11.7%), and the median tonic-atonic seizure frequency as compared to placebo (42.5% vs. −1.4%).54 In addition, subjects or their parent/guardian in the RFM treated group noted a significant improvement in seizure severity as compared to the placebo group (53.4% vs.30.6%).54 The proposed target dosage of RFM (45 mg/kg per day) was achieved by 87.8% of the RFM treated subjects.54
Although RFM has not been approved for partial epilepsy, a randomized, placebo-controlled study was conducted in 357 subjects aged 16–72 years. RFM was initiated at 800 mg/day in two divided doses (400 mg BID), with a maximum dosage of 3,200 mg/day. The study demonstrated that RFM reduced the median partial seizure frequency significantly (23.3% vs. 9.8%).55
RFM appears to be well tolerated in long-term use. Of the 139 subjects who completed a multicenter, double-blind, placebo-controlled, randomized study of RFM as adjunctive treatment for seizures associated with LGS, 124 continued into an open label extension phase with a median treatment period of 432 days. Only 12 of these subjects ultimately discontinued the medication due to adverse side effects.56 The most commonly reported TEAEs were dizziness, fatigue, nausea, vomiting, diplopia and somnolence. In LGS clinical trials, the most commonly reported reason for withdrawal of RFM treatment in adult patients was dizziness.54 RFM has been shown to be associated with shortening of the QT interval (up to 20 msec) on EKG studies as compared to placebo. RFM’s effect on QT interval was proportional to serum concentrations. Although it was not clear whether or not the patients were symptomatic from RFM’s effect on the QT interval, there has been no drug-induced sudden death or ventricular arrhythmias associated with its treatment. The clinical risk associated with RFM -induced QT interval shortening is unknown. However, caution should also be used when RFM is given in conjunction with other medications that may shorten the QT interval such as ranolazine, primidone, or lamotrigine.57 RFM is contraindicated in patients with Familial Short QT syndrome.51
Perampanel
Perampanel (PRP) is potentially a new anti-epileptic compound that has unique MOA and structurally distinct from other AEDs. Although it is a new compound, considerable clinical experience derives from previous trials of PRP for Parkinson’s disease. PRP has not yet been granted marketing authorization by the EMEA or the FDA even though it was submitted to the FDA for approval as an adjunctive therapy for partial onset seizures.
Pharmacokinetics
PRP is rapidly absorbed following oral administration, with peak plasma concentrations reach between 15 min and 2 hours. The half life is estimated to be about 70 hours, which allows once a day dosing, and the steady-state plasma concentration of PRP is achieved in 14 days. The distribution volume is about 77 L with high protein binding of 95%.58 Following oral administration of radiolabled PRP, 70% was excreted via feces in unchanged form, and remaining 30% through kidney.58 Unchanged PRP is found circulating in the serum, while glucuronidated and hydroxylated metabolites are found in urine.58 In vitro studies indicated that the CYP 3A4 pathway is the primary route of CYP mediated metabolism of PRP.58
PRP does not affect the plasma concentration of CYP-inducing and non-CYP inducing AEDs, such as carbamazepine, topiramate, oxcarbazepine, levetiracetam, lamotrigine or valproic acid. However, perampanel concentrations were noted to be 50% lower when used with carbamazepine.58 The impact of perampanel on the effectiveness of oral contraceptives has not been studied.
Mechanisms of action
PRP is structurally very distinct from other currently available anti-epileptic medications. PRP is highly selective antagonist of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and in cultured cerebral cortical neurons from embryonic rats, PRP was shown to inhibit the AMPA induced increase in intracellular calcium concentration, thereby reducing excessive excitatory responses.58
Efficacy and tolerability
Clinical efficacy and safety data are available from two Phase II and one recent phase III studies. The phase II studies evaluated PRP dose of 4, 6, 8, 10, and 12 mg/day and randomized 153 patients. The responder rates over the 4-week maintenance treatment phase were ranged from 31–40% on PRP compare to 20–22% on placebo. Median reduction in seizure frequency was similar but neither study was sufficiently powered to detect statistical significance of these results. A phase III, multi-center, randomized, double-blind, placebo controlled, dose escalation, parallel group study performed primarily in the United States, Europe and Asia (total of 25 countries) was conducted to study the efficacy of PRP as adjunctive therapy for the treatment of refractory partial seizures. The study enrolled 712 subjects with partial seizures with or without secondary generalization, ages 12–72 years old. Of the enrolled subjects, 623 completed the trial. Subjects were randomized to PRP 2, 4, or 8 mg/day or placebo. Patients could be concomitantly treated with 1–3 anti-epileptic medications with a maximum of 1 inducer medication. The study demonstrated statistically significant reduction in seizure frequency with use of PRP.58
In the phase II studies, the most common adverse events occurring in more than 10% of any treatment group were dizziness, somnolence, headache, fatigue, diarrhea, rhinitis, nasopharyngitis, and convulsions.58 Most of the events were mild and not dose limiting. In phase III studies, withdrawal due to adverse events was low in all treatment groups and in the placebo arm.59 In phase I and II studies, no life threatening adverse events were noted.60 There were no clinically significant changes reported in physical examinations, vital signs, laboratory testing or electrocardiograms.60
Deep brain stimulation
Recent theories in the propagation and circuitry of epilepsy and seizures have led to interest in direct brain stimulation as a potential treatment modality in epilepsy. Deep Brain Stimulation (DBS) has proven to be effective for treatment of movement disorders over time, in particular Parkinson’s disease and essential tremor. Targeted areas of stimulation in those disorders are commonly the subthalamic nucleus, the globus pallidus pars interna, and the thalamus.61 The utility and anatomical targets of DBS in other disorders were evaluated, and small studies had shown promise in DBS treating epilepsy by stimulating various sites. A previous study suggests that DBS stimulation of the anterior thalamus directly affects the ipsilateral hippocampus and mesial temporal lobe.62 DBS device was approved by the EMEA recently for the treatment of refractory partial seizures, but it s not yet approved by the FDA.
Efficacy
Based on these small studies, Stimulation of the Anterior Nucleus of the Thalamus in Epilepsy (SANTE) study had been completed. This study was a prospective randomized, double-blind, multicenter trial, which enrolled 110 people with refractory partial-onset seizures.63 All patients had surgery with DBS electrode placement in the anterior nuclei of the thalamus, as evidenced by post-operative MRI. One month after surgery, half of the participants had their DBS turned on at 5V, the other half had the stimulation maintained in off mode at 0V, in a blinded manner. Parameters for stimulation were 145 pulses per second, “on” for 1 min, and “off” for 5 min. After this 3 month blinded phase, all patients underwent DBS from months 4 to 13 at predetermined parameters. Results were favorable, with a 29% greater reduction in seizures in the treated group compared with the control group during the 3 month blinded period. There was a 40.4% reduction in seizures in the treated group compared to 14.5% in the untreated group at month three after randomization (p=0.0023). The prior months showed a trend towards more seizure reduction in the stimulation group, but significance was not seen. Severity of seizures and injuries from seizures were also statistically reduced in the treatment group compared to control.63
Subgroup analysis showed a difference in response to treatment dependent on location of seizure origin. Those with temporal lobe onset had a median seizure reduction of 44.2% in the treatment group compared to a 21.8% reduction in subjects receiving control treatment (p=0.025). However, there was no significant difference in seizure reduction between treatment and control groups in patients with frontal, parietal, or occipital originating seizures. Patients with a vagus nerve stimulator and those with previous respective surgeries were included and had similar results to other patients in the study. At 13 months during open label phase, there was a 41% seizure reduction, and at 25 months, there was a 56% reduction in seizures (n=99). At 13 months, the 50% responder rate was 43%, at 25 months, 54%, and at 37 months, 67%.63 This data suggests that long-term therapy may be necessary for significant improvement, being a delayed effect in some patients.
Safety and tolerability
Risks in DBS surgery include hemorrhage, infection, and psychological side effects. The most serious risk is local hemorrhage in the site of probe placement. Incorrect probe placement can also be an issue, especially when targeting very selective areas such as the centromedial thalamus, or the subthalamic nucleus. Infection can occur- either intracranial during the surgery, or surrounding the device and leads extracranially. Some studies in Parkinson’s patients have noted depression or other neuropsychiatric side effects after DBS,64 and guidelines have been established that patients with active depression or psychiatric illness are generally excluded from DBS in that group of patients. There is an overall 1–3% risk of serious side effects from placement of DBS.
In the first 13 months, 808 TEAEs were reported in the study group. Fifty-five events were considered serious. Depression and memory impairment were the most common side effects in the treatment group. Paresthesias, implant site pain, and implant site infection (9.1%), were the most common device related events. Although implantation related hemorrhagic complication had occurred, none of them were reported to be symptomatic. Total of 16.4% of patients withdrew from the study because of adverse events. There were five deaths in the study although none judged to be related to the device.63,65
Responsive neurostimulation
Recent investigations have been completed in a closed-loop circuit electrical stimulation for the treatment of refractory epilepsy. In responsive stimulation systems, in addition to the stimulating electrode, a receptive electrode is placed just beneath the dura, or as a depth electrode near the epileptic focus. This electrode measures for epileptogenic activity. If epileptogenic discharges are detected in this receptive electrode, a high-frequency stimulation is sent via the stimulating electrode, theoretically aborting the seizure. Data from the device can be downloaded into a computer via a hand-held wand and settings can be adjusted for the receptive and stimulating electrodes. RNS has not been approved neither by the EMEA nor the FDA yet for the treatment of seizures.
Efficacy and safety
A randomized sham control trial for safety and effectiveness of the responsive neurostimulator (RNS) system had been completed. Subjects had localization-related epilepsy and had 3 or more seizures per month (ages 17–80), and had failed at least 2 AEDs. A total of 191 adult patients were enrolled. The treatment group had the system turned “on” for three months while the sham group’s RNS remained off. After the three month blinded period, all patients were treated with active RNS systems. During the blinded portion of the study, reduction in seizure frequency was 37.9 percent in the treatment group and 17.3 percent in the control group (p=0.012). For all patients actively treated two years, there was 50 percent responder rate of 53% of total patients.66
Depression and subjective memory complaints were the same in the sham and treatment groups.66 There were no serious device side effects reported although the open label portion of the trial is still on-going.
Discussion
There are increasing numbers of new AEDs introduced within the last 5 years and more than a dozen of medications are in pipeline. When a new treatment of epilepsy AED is introduced, clinician often wonders what are unique and new features of the medication. Therefore, understanding the differences in pharmacokinetics, MOA, potential drug-to-drug interactions, and tolerability may provide useful guidance when choosing a new AED for epilepsy patients. Favored AEDs should have reliable bioavailability, linear kinetics, low or no drug-to-drug interactions, low protein binding, longer half-life, convenient dosing, and preferably a novel MOA. Although not all compounds display favorable properties in all of these regards, there are some notable advantages over existing AEDs. For example, many of these AEDs have novel MOAs with clean pharmacokinetic pro perties (Table 2). Unfortunately, despite the fact that there are new properties of new AEDs, overall reported efficacy seems be similar to each other and that of existing AEDs. Beside efficacy, perhaps the more important question is whether these new medications could improve the quality of lives of patients with epilepsy, which are not usually answered by conventional clinical study design for epilepsy.
Although these new AEDs provide unique and novel MOAs for seizure treatment, it is not clear to what extent and how theses differences in MOAs would impact clinical practice. For example, the benefit of combining AEDs with different MOAs still remains unclear. When combination therapy is considered, using AEDs with different MOAs may provide better efficacy and tolerability, and even possibly a synergistic (supra-additive) effect. On the other hand, using AEDs with similar MOAs may result in simple additive or even antagonistic (infraadditive) effect. However, previous clinical studies had failed to demonstrate that combination of similar MOAs are inferior to combination of different MOAs. For example, the efficacy of ESL was not different whether it was added to sodium channel blocking AEDs or not. More recently, isobolographic analysis in rodent models has been used to determine whether combination of two medications could be synergistic, additive, or antagonistic to each other.67 Nonetheless, it is not yet clear how these combinations would impact the seizure control in clinical practice. To date, there are no clinical studies examining the pharmacodynamic interactions or clinical efficacy of these medications with other existing AEDs.
The new modalities of direct brain stimulation are certainly intriguing. Since 1997, many patients have been benefited from vagus nerve stimulation (VNS) therapy There are several main differences between VNS, DBS, and RNS. First of all, VNS is considered as indirect stimulation of brain via intermittently stimulating peripheral vagus nerve. While DBS is targeted the anterior thalami, RNS is directly targeted the epileptic zone. Second, RNS is the first close loop approach which can monitor potential seizure activity and deliver electrical stimulation on demand. In contrast, VNS and DBS provide periodic stimulation around the clock without monitoring seizure activity. Third, generator and battery packs are implanted in the chest area while generator of RNS is embedded in the superficial portion of the patient’s skull. Fourth, side effects from VNS can be readily felt by patients (i.e. coughing, and voice changes) but most of patients cannot immediately sense the stimulation with DBS or RNS. And the fifth, complexity of programming the devices and patient selection may impose challenges for RNS and DBS compare to simple steps in VNS programming. Overall, when these devices are utilized for properly selected epilepsy patients, they can provide significant improvement without exposing them to common side effects of AEDs. However, it should be pointed out that not all patients would be benefited from these electrical stimulations even after careful patient selection just like medication therapy. These are currently adjunctive treatments that will require further studies to evaluate the most appropriate modalities of stimulation (i.e. frequency, intensity, duration, and the location of stimulations).
Conclusion
There are many new generation medications with favorable pharmacokinetics and potentially novel MOAs, and new modalities of electrical brain stimulation for the treatment of epilepsy. Results from clinical studies demonstrate that many of these AEDs and devices were well tolerated and effective in reducing partial onset seizures as adjunctive therapy. The availability of these new treatments for epilepsy will expand treatment options for patients and may provide a significant benefit to some patients who remain refractory to existing therapy.