Introduction
Neurofibromatosis is well known neurological inherited disorder, which is characterized by café-au lait macules, Lisch nodules, neurofibromas, and learning disabilities.1 The prevalence of neurofibromatosis type I is about 1/3,000 individual.2 It is an autosomal dominant neurocutaneous condition. The mutations are located in chromosome 17q11.2, which codes for neurofibromin (NF1) and detected in about 95% of patients.3 Abnormal NF1 gene may be linked to malformations of cortical development, such as dysembryoplastic neurepithelial tumors, gangliogliomas, and unspecified cortical malformations.4
Neurofibromatosis type 1 is commonly combined with various types of malformations, which include hemimegalencephaly, cerebellar leptomeningeal heterotopias, transmantal cortical dysplasia, periventricular band heterotopias, pachygyria, occipital encephalocele and unilateral as well as bilateral polymicrogyria.5 It is one of the neurocutaneous syndromes, which has commonly epilepsy as a common clinical feature. But epileptic seizures are not common clinical manifestations unlike other neurocutaneous syndrome. Epilepsy occurs in about 4–7% of individuals and seems to be related to cortical dysplasia.6,7 Most of seizures are mild and well controlled,6 while coexistence of other malformations of cortical development is usually accompanied by pharmacoresistent epilepsy syndrome.
We report a patient with neurofibromatosis type 1 who showed intractable mesial temporal lobe epilepsy.
Case report
A 32-year-old lady had developed seizures 5 years before visiting our hospital. She was a right handed postgraduate student. Her mom reported that she fumbled her fingers and showed lip smacking during her seizures and did not respond to questions. She reported only one episode of secondarily generalized seizures, which occurred before taking any antiepileptic drugs. The auras were the most frequently abdominal, but rarely vertiginous or psychic. The initial brain MRI had showed unusual multifocal lesions. Stereotaxic biopsy did not confirm the etiologic diagnosis.
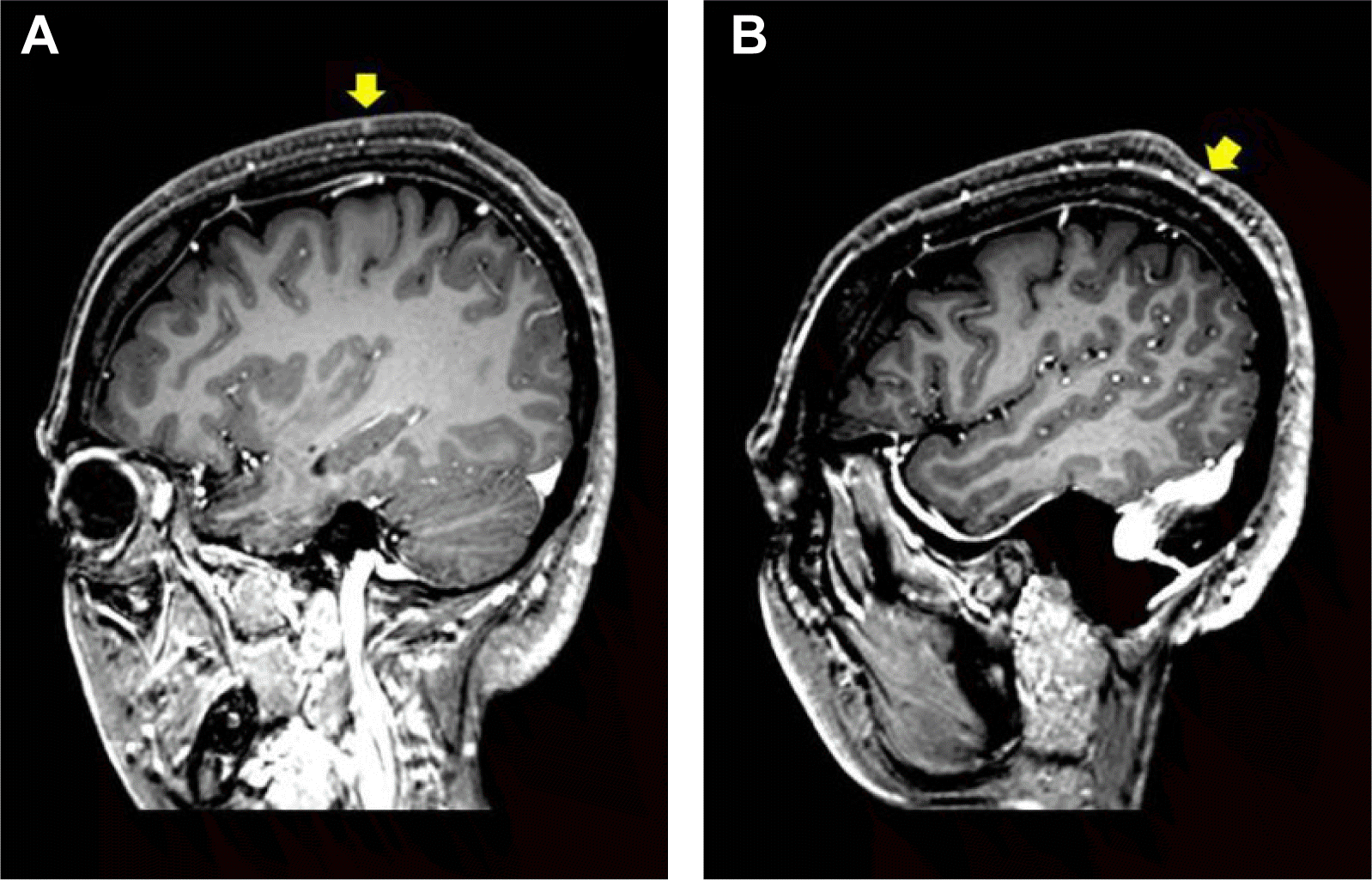
After the biopsy, she had been transferred and followed-up at our epilepsy outpatient clinic for seizure control. On family history, her grandmother had seizures. Physical examination showed apparent axillary pigmentation and freckling, seven café-au-lait spots which are more than 15 mm on back. Neurological examination, including visual, and auditory and somatosensory systems, did not reveal any focal deficit. Serial brain MRI showed multifocal lesions in right temporal area, left tectum, right cerebellar peduncle thalamus, basal ganglion, and intraventricular cyst in left trigon of the ventricle (Fig. 1). Those lesions were well localized and not enhanced, and were not accompanied by surrounding edema, which were compatible with CNS hamartomas. The right mesial temporal area showed increased signal changes and increased volume, prominently in the head of hippocampus. The volumetric study of hippocampi demonstrated that the total volume of right hippocampus was slightly bigger than that of left hippocampus [right hippocampus: 4,105 mm3 (normal range: 2,563–4,195 mm3), left hippocampus 3,659 mm3 (normal range: 2,476–3,973 mm3)]. The difference of right to left hippocampal volume was 446.14 mm3, which was in the normal range (−38.3–531.3 mm3). Those lesions were suggestive of multifocal infiltrative glioma, pleomorphic xanthoastrocytoma with multifocal involvement, other CNS lymphoma, vasculitis, and etc. However the enhanced MRI demonstrated multiple small lesions mainly in the scalp areas, indicating neurofibromas in skin, even though those lesions did not show clinical features (Fig. 2). Those skin manifestations and scalp MRI findings suggested neurofibromatosis type 1.
After the polytherapy of antiepileptic drugs, the intensity of her seizures was milder, but the frequency of her seizures was not changed. During the hospital course, she had simple partial seizures monthly and, complex partial seizures twice per year, when she had been on oxcarbazepine 1,500 mg, topiramate 400 mg, levetiracetam 3,000 mg, clobazam 10 mg, and pregabalin 150 mg per day.
Intellectual assessment revealed that the score of full scale IQ was 85, that of verbal IQ score 87, and that of performance IQ 84 which was estimated to be between 14.2–19.2 percentile. Her neuropsychological tests showed diffuse cerebral dysfunction. Beck’s depression inventory score was normal.
For treating intractable seizures and obtaining the pathologic tissues, presurgical evaluation of epilepsy surgery was performed, including video-EEG monitoring, Wada test, and detailed neuro-imaging studies.
She underwent three day video-EEG monitoring. During the stay, 10 complex partial seizures and 1 simple partial seizure were recorded. The one simple partial seizure was only abdominal aura. The complex partial seizures were consisted of abdominal aura, lip smacking, leaning backward, often with right or both hand fumbling, but, not accompanied by generalized seizures.
The interictal epileptiform discharges were frequently seen on right mid-temporal area about once per minute. The 11 ictal epileptiform discharges started with semi-rhythmic theta waves in right temporal area, and then delta waves in the same area, spreading into whole leads (Fig. 3A).
She underwent ictal and interictal 99m-Tc HMPAO SPECT. The radiotracer for ictal SPECT was injected at 40 seconds from the onset of 102 second duration habitual complex partial seizure. The hyperperfusion was localized to right anterior mesial temporal area. Interictal SPECT, taken after 24 hour seizure freedom, showed focal hypoperfusion in right mesial temporal area. Subtracted ictal SPECT co-registered to MRI demonstrated definitely hyperperfused areas in right mesial temporal regions, which are concordant to the localization of ictal EEG onset (Fig. 3B). Other lesions did not show any perfusion changes in SPECT studies. Brain 18F-FDG PET showed right temporal hypometabolism. Other brain and scalp lesions, which were confirmed on brain MRI, did not reveal any metabolic changes in PET study. Wada test demonstrated that her left hemisphere were both dominant for speech and memory, predicting good surgical outcome of right anteromesiotemporal resection.
After the removal of right anteromesial temporal area, her seizures became free. The pathologic tests showed mild neuronal loss at hippocampus, and dysplastic changes in the specimen of lesion tissue without any evidence of neoplasm.
Discussion
This is the first case report on an epilepsy surgery in a patient with pharmacoresistant mesial temporal lobe epilepsy due to neurofibromatosis type 1. Clinical diagnosis of neurofibromatosis type I requires 2 or more of the following: first, 6 or more café-au-lait spots (>5 mm in diameter for prepubertal children and >15 mm in postpubertal patients); second, a plexiform neurofibroma (or 2 or more neurofibromas of any type); third, Crowe sign (multiple freckles in the axillary or inguinal region); fourth, a first degree relative with confirmed neurofibromatosis type 1; fifth, optic nerve glioma; sixth, 2 or more Lisch nodules (hamartomas); seventh, sphenoid dysplasia, cortical thinning in long bones, or other distinctive osseous lesion.8,9 In our cases, she had the first criteria of café-au-lait spots and the third criteria of axillary freckles. Also the second criteria of plexiform neurofibroma might be on her because of scalp lesions on brain MRI.
The brain MRI findings show multiple cerebral, cerebellar and brainstem lesions. If we considered the pattern of involved lesions, the tectum, cerebellar peduncle, thalamus, basal ganglia, and mesial temporal area are near the center, indicating paramedian arrangement of lesions in brain. Also the findings of scalp on brain MRI demonstrate enhanced multiple focal subcutaneous or cutaneous lesions. The scalp lesions could be differentiated from the vessels, if we look into the adjacent images. The vascular structures show the continuous structures on the adjacent images. Usual brain MRI has high signal intensities on the scalp due to fat. So fat suppression imaging technique are needed, which is commonly applied in FLAIR image. Also small neurofibromas can be omitted by large slice thickness. So the thickness of section should be as thin as possible. Also the possibility of nonspecific nodules could be differentiated by enhancement. The neurofibromas in neurofibromatosis type 1 are easily enhanced in the homogenous pattern. In the patient’s thin slice, fat suppression, enhanced brain MRI, the enhanced focal scalp lesions are indicative of neurofibromas, and brain lesions are indicative of paramedian arrangement.
Malformation of cortical development, including cortical dysplasias, is one of the epileptogenic brain lesions.1,10 One of them was the cause of epileptogenic focus. After removing the lesion, the patient became seizure free. The lesion was located in mesial temporal area, suggesting that neurofibromatosis type 1 can be one of the causes of mesial temporal lobe epilepsy. The clinical characteristics of mesial temporal lobe epilepsy in neurofibromatosis type 1 are similar to those in hippocampal sclerosis except for the febrile seizure. Moreover the pathology of our case showed mild neuronal loss, suggesting the secondary epileptogenesis, and mild focal cortical dysplasias as well. Those findings may support that neurofibromatosis type 1 could be one of the causes in mesial temporal lobe epilepsy. The hippocampal neuronal loss and mid focal cortical dysplasia in mesial temporal structure can be pathologic findings of neurofibromatosis type 1 as an epileptogenic focus.
In most neurofibromatosis type 1 patients with seizures, lesionectomy can be performed effectively if a single and well-delimited epileptogenic zone is recognized.1 Conversely, MRI evidence of other malformations of cortical development such as focal cortical dysplasia and polymicrogyria is usually accompanied by drug-resistant epilepsy.4,10
We report a case with pharmacoresistant mesial temporal lobe epilepsy with neurofibromatosis type 1, in which the anteromesial temporal resection worked effectively, leading to seizure-freedom.
The dysplastic lesion of mesial temporal lobe in a patient with NF1 may be epileptogenic and become pharmacoresistant. In such a case, early diagnosis and surgical removal is necessary to make the patient become seizure-free. Presurgical evaluation including video-EEG monitoring, MRI, SPECT, PET and Wada test is mandatory for the precise localization of epileptogenic foci.