Introduction
Carbon monoxide (CO) poisoning is a medical emergency that leads to cardiovascular and neurological complications.1 The clinical manifestations of CO poisoning are nonspecific, and various neurologic abnormalities, such as confusion, seizures, and coma, may occur at higher levels of carboxyhemoglobin (COHb).1 Various reports show self-limiting seizures, but status epilepticus has rarely been reported in patients with CO poisoning.1,2 Furthermore, hyperbaric oxygen therapy (HBO2) may result in late-onset neurological sequelae.3 The exact pathogenesis underlying HBO2 and status epilepticus associated with CO poisoning is not yet clear. Herein, we present a patient who developed non-convulsive status epilepticus after acute CO poisoning treated with HBO2 therapy.
Case
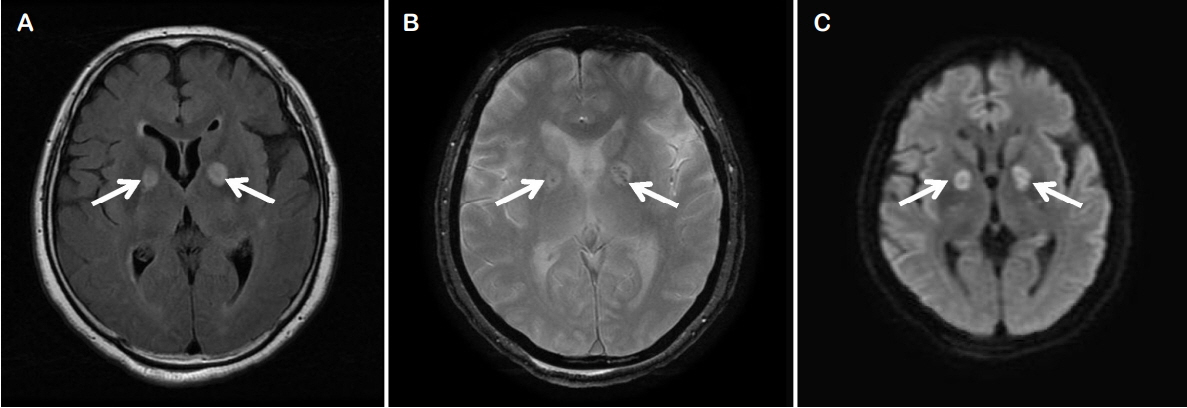
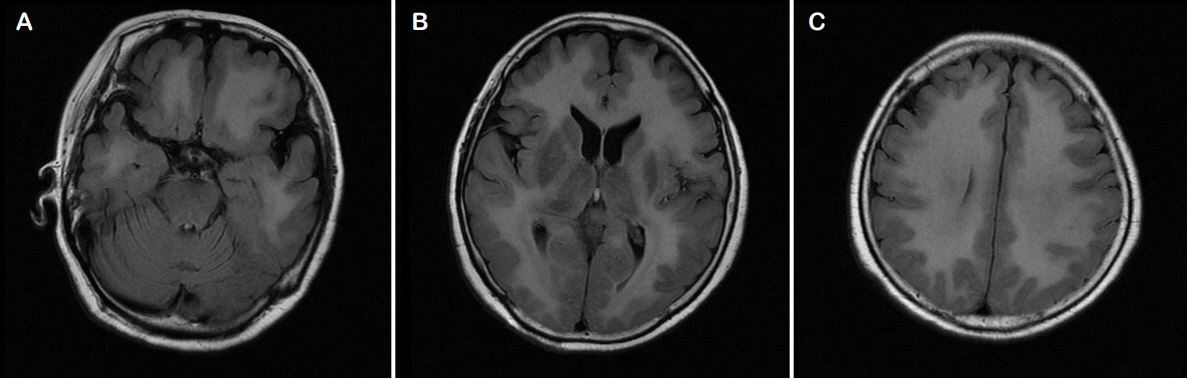
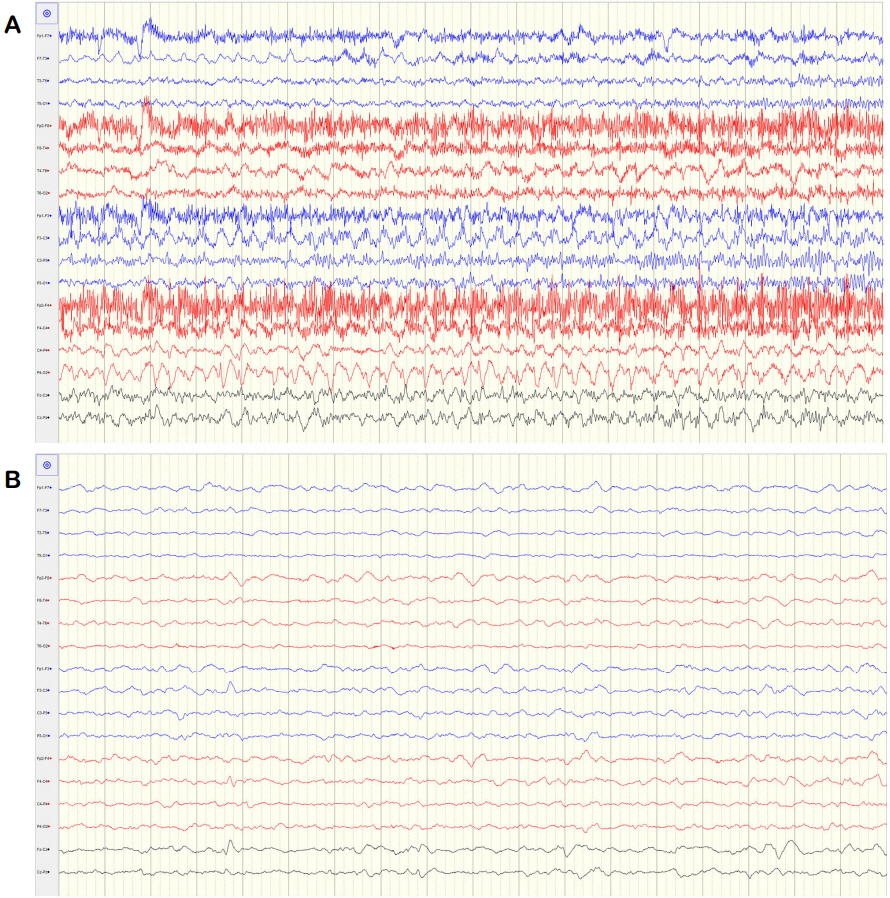
A 71-year-old woman attempted suicide by burning charcoal at home. She was found unconscious after 3 hours of exposure to CO from the burning charcoal. Apart from burning charcoal, she also took an unknown amount of hypnotic drugs. She had been treated with risperidone, amitriptyline, and escitalopram for paranoid schizophrenia and bipolar disorder during 10 years. On arrival at our emergency room, she was unconscious with a Glasgow coma scale (GCS) score of 7/15. The pupils were bilaterally equal, 3 mm in size and reactive to light. Examination of the vital signs found an oral body temperature of 37°C, a regular pulse of 93 beats/min, blood pressure of 67/45 mmHg, and pulse oximetry of 93% in room air. Her arterial blood gas results in room air were as follows: pH 7.48, PaCO2 32 mmHg, PaO2 50 mmHg, base excess −2.1 mmol/L, SaO2 95.2%, and COHb 19.3%. Laboratory tests found a serum prolactin level of 69.19 ng/mL, high sensitivity-C reactive protein (hs-CRP) level of 1.719 mg/dL, and erythrocyte sedimentation rate (ESR) of 15 mm/h. No abnormalities were detected in the cerebrospinal fluid except for mildly elevated protein contents of 51 mg/dL. Urine toxicology screening tests gave negative results. Other results of blood chemistry was normal. Electrocardiography revealed sinus tachycardia without a change in the ST segment. Brain magnetic resonance imaging showed bilaterally symmetric focal abnormalities involving the globus pallidi (Fig. 1). CO poisoning was suspected because of the increased level of COHb (19.3%) and 100% oxygen administration was initiated. She underwent three HBO2 sessions over the course of 3 days. Following HBO2 therapy, the patient recovered spontaneous respiration and the tube was removed. On neurological examination, she was found to be alert and able to communicate normally (GCS 13). On day 28, she was unconscious with a GCS score of 3/15 and her COHb level was 0.9%. Magnetic resonance imaging revealed a diffuse, confluent hyperintensity on T2-weighted sequences in bilateral cerebral white matter, suggesting demyelination of the cerebral white matter (Fig. 2). An electroencephalogram (EEG) was performed and showed continuous and diffuse high amplitude paroxysmal sharp waves at 2–3 Hz (Fig. 3A). She was diagnosed with non-convulsive status epilepticus and intravenous infusion of 1,000 mg of levetiracetam and 1,500 mg of valproic acid was therefore administered. Continuous EEG monitoring showed an absence of epileptiform activity (Fig. 3B). On day 40 after CO poisoning, she was semi-comatose with rare responses to painful stimuli and transferred to a nursing hospital without any recovery (GCS 3).
Discussion
We experienced a patient with CO poisoning with non-convulsive status epilepticus treated with HBO2. CO is an odorless, colorless, and nonirritant gas produced by the incomplete combustion of carbon-based fuels. The clinical features of CO poisoning are non-specific, and physicians may consider a broad range of diagnostic possibilities.
In the present case, several differential diagnoses can be considered as potential causes of mental deterioration with non-convulsive status epilepticus. Firstly, past medical history of the patient revealed that she had been under treatment with risperidone, amitriptyline, and escitalopram for paranoid schizophrenia and bipolar disorder, which can give rise to drug intoxication resulting in seizures. Acute drug intoxication is a condition that follows the administration of a psychoactive substance and results in disturbances in the level of consciousness, cognition, perception, judgement, or seizure, or other psychophysiological functions and responses. Antidepressant drugs, such as amitriptyline and escitalopram, and atypical antipsychotic medication are known to have a proconvulsive effect.4 However, in this case, drug intoxication induced seizure seemed highly unlikely because she took usual drug dose and her urine toxicology tests were normal. Second, metabolic causes of acute altered mental status must also be ruled out and are common among the elderly with underlying medical conditions. Metabolic causes of altered mental status include hypo/hyperglycemia, electrolyte abnormalities, uremia, hepatic encephalopathy, hypertensive encephalopathy, metabolic encephalopathy, and hypo/hyperthyroidism.5 Acute metabolic encephalopathy is a condition of global cerebral dysfunction in the absence of primary structural brain disease.
However, in our case, her blood chemistry was normal except for elevated prolactin, hs-CRP, and ESR level and brain magnetic resonance imaging showed bilaterally symmetric focal lesions involving the globus pallidi. Finally, while central nervous system (CNS) infection can also induce status epilepticus, the results of cerebrospinal fluid analyses were incompatible with the presence of infection. In this particular case, the patient was discovered after 3 hours of exposure to the charcoal fumes. These circumstances may lead to CO intoxication and consecutive non-convulsive status epilepticus.
In the early stages of CO poisoning, most patients present with various neurologic complications, including apathy, amnesia, disorientation, extra-pyramidal symptoms, psychosis, seizure, and coma.6 In adults, mild symptoms, such as headache, dizziness, palpitation, and nausea, are typical in patients with COHb levels of 10–30%.6 Seizures are generally thought to occur when the COHb level reaches more than 40%, but the level does not necessarily correlate with the severity of symptoms.7 In our experience, the COHb level was 0.9% when the patient was diagnosed with non-convulsive status epilepticus. There may be possible hypotheses regarding whether her low COHb level was related to non-convulsive status epilepticus in the late stages of CO poisoning. The pathophysiology of this delayed and repeated seizures depends on various mechanisms. CO has an affinity for hemoglobin 200–250 times greater than that of oxygen, forming COHb and reducing the total capacity of the blood to carry oxygen. This results in cerebral hypoxia far out of proportion from what would be expected from the loss of oxygen-carrying capacity. CO causes mitochondrial dysfunction resulting into oxidative stress related damage with elevated levels of excitatory amino acids (N-methyl-d-aspartate) and brain nitrites.8 With reperfusion of the brain, leukocyte adhesion and the release of destructive enzymes amplify the initial oxidative injury.8 This delayed seizures may occur even when COHb level were low. Furthermore, cellular theories such as immunopathological damages and disturbance of dopaminergic and serotonergic functions support the underlying pathophysiology of this delayed seizures. This theory postulates that the delayed but reversible effects were related to the activation of polymorphonuclear leukocytes by the CO, leading to reversible demyelination of the CNS.9 In the meanwhile, elderly patients may be at high risk of CO poisoning because of a slower elimination rate of CO than pediatric patients. The elderly has an increased neurologic susceptibility to CO poisoning and may experience CNS symptoms at a much lower dose.10 In this case, the patient has higher risk for CO poisoning due to her old age.
We treated a patient with non-convulsive status epilepticus without self-limiting isolated seizures after exposure to repeated HBO2. There are many case reports and retrospective studies demonstrating seizures induced by HBO2 therapy.11 HBO2 is the most effective treatment for patients with acute CO poisoning. The underlying mechanism includes increasing dissolved-oxygen content in the blood and accelerated elimination of CO, with prevention of lipid peroxidation and preservation of adenosine triphosphate (ATP) levels in tissue in serious CO poisoning.12
CNS oxygen toxicity is a rare but recognized side effect of HBO2.13 Our case presented non-convulsive status epilepticus after treated with HBO2. The pathogenesis of seizures associated with HBO2 is not fully understood but may be associated with the decrease in the seizure threshold through various mechanisms. Firstly, in CO poisoning treated with HBO2, oxidative stress develops during the ischemic and reperfusion phases and increases the production of nitric oxide (NO) by NO synthase. Increased NO levels inhibit glutamic acid de-carboxylase and results in a decrease in gamma-aminobutyric acid levels.14 NO can also have toxic effects by inhibiting aconitase, complex I and II in the electron transport chain, and mitochondrial respiration, resulting in ATP depletion and disruption of the active transport system of the cell.15 NO might cause toxicity by increasing intracellular free calcium by activating the ryanodine receptor. These changes can induce the activation of a series of enzymes and disruption of the cell membrane potential, which might result in seizures.15 In mice exposed to HBO2, these changes have been observed in the brain stem, corpus callosum, and cerebral cortex.16 Second, brain-derived neurotrophic factor (BDNF), as a survival factor for certain neuronal population, provoked by HBO2 therapy might modify both excitatory and inhibitory synaptic transmission in the brain.17 The increased BDNF signaling plays a role in the development of seizure and epileptogenesis. Finally, HBO2-induced changes in cerebral blood flow (CBF) may play a important role in repeated seizure generation.18 Increased CBF during seizure might represent epileptogenic zone. In normal conditions, CBF is regulated by the level of carbon dioxide. However, with prolonged HBO2, cerebral vaso-dilation might occur and produce free radicals and nitric oxide, predisposing to more convulsions.18
Radiologic findings in CO poisoning are variable. During the acute phase of CO poisoning, the globus pallidus may be one of the most frequently injured areas.19 In delayed encephalopathy, the most common finding is white matter hyperintensity that might be caused by demyelination after CO poisoning. Sometimes, lesions may be found in the hippocampus or cortex that are correlated with a poor prognosis in the delayed phase of CO poisoning.19 The MR image from our case showed the same involvement and abnormal signal intensity of bilateral basal ganglia in the acute phase and diffuse white matter in the delayed phase. During the early phase of CO poisoning, to better correlate the symptoms with the clinical course, imaging techniques, such as diffusion tensor imaging or diffusion-weighted MRI, may be helpful in detecting white matter lesions.20
In conclusion, non-convulsive status epilepticus is rare both after CO poisoning and HBO2 therapy. Our case demonstrates that clinicians should need to be aware of delayed and fatal complications, such as non-convulsive status epilepticus and perform immediate EEG and MRI when mental status fails to improve after HBO2 therapy in CO poisoning.