Introduction
Temporal lobe epilepsy (TLE) is associated with oxidative stress and neuronal apoptosis.1 Antiepileptic drugs can control seizures but also appear to promote oxidative stress and apoptosis.2 One of the consequences of insufficient antioxidant and anti-apoptotic activity is loss of neuronal interconnectivity within the brain’s learning and memory networks.2 Neuronal regeneration has been seen in experimental models of temporal lobe epilepsy.3–6 However, whether neuronal regeneration is linked to attenuation of the cognitive deficits and neurobehavioral symptoms of TLE is largely unknown. Cognitive challenges, aerobic exercise and various forms of physical activity have been shown to improve clinical outcomes following physical brain injury.7–9 Such interventions are considered forms of environmental enrichment (EE).10–13 The present study was, therefore, designed to test the hypothesis that EE increases neurogenesis at sites of motor, learning, memory and behavioral regulation and attenuates the learning and memory deficits associated with seizures in an animal model of TLE.
Methods
This research was approved by the Ethical Clearance Committee of Manipal University.
Animals
Male Wistar rats (4 month-old) were used. All the cages were maintained in a 12-h light and 12-h dark cycle in well-ventilated rooms within the Manipal University Animal House. All rats were fed ad libitum with a balanced diet containing 21.96% crude oil, 3.10% crude fiber, 7.37% ash, 1.38% sand silica.
Experimental design
Rats were divided into five groups 1) normal control, 2) normal control + exposure to EE, 3) sham control + exposure to EE, 4) Kainic acid-lesioned, and 5) Kainic acid lesioned + exposure to EE. Rats in the normal control group remained undisturbed in the home cage. Rats in the normal control + EE group were subjected to EE for 3 h/day. Rats in the sham control group were subjected to sham surgery. The sham surgery consisted of positioning the rats in a stereotaxic apparatus, burr holes were then drilled in the skull using appropriate coordinates. A Hamilton syringe was lowered into the lateral ventricles bilaterally and removed. The scalp wounds were sutured, animals were then replaced back in their home cage and subjected to EE. Rats in the lesion only group were administered kainic acid (KA) bilaterally into the hippocampus using a Hamilton syringe. Rats in the KA lesioned + EE group were lesioned with KA and were subjected to EE for 3 h/day.
Experimental procedures
An excitotoxic lesion was created in the hippocampus by injecting KA into the lateral ventricles using established stereotaxic coordinates.14 Rats were first anaesthetized with a cocktail of ketamine (50 mg/mL), xylazine (4.5 mg/mL) and acepromazine (0.4 mg/mL) at a dose of 0.70 mL/kg body weight and were fixed in the stereotaxic apparatus so that the incisor bar was 3.7 mm below the interaural plane. The skull was exposed, and a burr hole was drilled using the following coordinates on the right and left sides: anteroposterior 3.7 mm behind the bregma 4.1 mm lateral to the midline.14 A Hamilton syringe needle filled with KA (0.5 μg/μL) was lowered by 4.5 mm to reach the lateral ventricle and 1.0 μL of KA was injected slowly over a period of 20 minutes. The needle was withdrawn, skin was sutured and the animals were kept warm until recovery from anesthesia. Lesioned animals were housed individually. Sham surgery was performed to rule out the effect of surgical injury. Here rats were anaesthetized, fixed in the stereotaxic apparatus and burr hole was drilled as described above. A Hamilton syringe needle was lowered and held in position for 20 minutes and then withdrawn. The skin was sutured and the animals were returned to their home cages.
EE
Exposure of animals to EE was carried out in a wooden cage of larger dimensions than the steel home cage. Each wooden cage contained different objects such as tubes, running wheel, ladder, and cubes etc., which were changed daily (Fig. 1). Rats were allowed to explore the enriched environment for 3 hours every day for 30 days, beginning immediately following either grouping (normal control group, sham operation [sham-operated control group] or kainate lesioning [kainate experimental group]) in the immediate exposure experiment and for 3 hours every day for 30 days beginning 60 days following the grouping into the sham operation or kainate lesioning in the delayed exposure experiment.
Morphological procedures
For the cresyl violet staining procedure, the animals were deeply anesthetized with ether and fixed on a dissection board and the chest cavity was opened to expose the heart. Fixation following transcardial perfusion was performed using about 15 mL of 0.9% heparinized saline perfused through the left ventricle at a rate of 1 mL/min. This was followed by perfusion with 10% formalin, about 250 mL/adult rat, at the same flow rate. The animals were decapitated and 5–6 mm thick coronal sections of brain were removed and kept in 10% formalin for 24 hours (post fixation). Paraffin blocks were made by tissue dehydration in 70% alcohol for 2 hours, 90% alcohol for 2 hours, three changes in 100% alcohol for 2 hours each, clearing with xylene for 2 hours and embedding using four changes of paraffin for 30 minutes each, followed by embedding in fresh filtered paraffin. Sections of 5 μm thickness were cut from the mid-dorsal hippocampus and motor cortex regions using a rotary microtome. Sections were selected and mounted serially on air-dried gelatinized slides. The sections were stained with cresyl violet, (0.1%) as follows: 100 mg of cresyl violet was dissolved in 100 mL of distilled water. To this, 0.5 mL of 10% acetic acid was added to a pH in the range of 3.5–3.8 log M. The stain was filtered before use. For staining sections were treated with two changes of xylene for 10 minutes, descending grades of alcohol (100%, 90%, and 70%) for 2 minutes each, distilled water for 15 minutes, 0.1% cresyl violet stain for 30 minutes at 60°C, cooling to room temperature, followed by treatments with ascending grades of 90% and 100% alcohol for 1–2 minutes each, xylene for 2 minutes, followed by mounting in DPX.
In each section CA1, CA3, dentate gyrus, amygdala and motor cortex regions were selected and the number of neurons counted using light microscopy.
Cell quantification
Total number of surviving neurons in the hippocampus (CA1, CA3, and dentate gyrus) and amygdala (basolateral nucleus) were counted. In the case of motor cortex, the number of surviving neurons in 10 randomly-selected 10 fields, at 40× magnification (Magnus, Olympus Pvt. Ltd., New Delhi, India), were counted and averaged. Cells that were darkly stained, shrunken with fragmented nuclei were excluded from the count. To avoid bias, slides from different groups were coded while counting.
Golgi-Cox staining
The Golgi-Cox staining procedure was followed with some modifications.15 Briefly, the rats were deeply anesthetized with ether and subsequently decapitated. The brains were quickly removed and placed in a petri dish containing freshly prepared Golgi-Cox fixative. The hippocampus was dissected from both hemispheres of the brain. The motor cortex was dissected and preserved separately. The caudal half of the other hemisphere with the hippocampus and amygdala region was collected for further processing. Tissue collected from individual animals was fixed in individual bottles as follows: brains were kept (as fresh as possible, without perfusion or fixation) on glass wool in clean bottles and covered with Golgi-Cox solution and left at room temperature in the dark room. The Golgi-Cox solution was refreshed after 2 days. Tissue was fixed for 2 weeks. After 2 weeks of impregnation in Golgi-Cox solution, the brains were processed further for dehydration in the following order: 50% ethanol for 1 hour, 70% ethanol for 1 hour, 90% ethanol for 2 hours, 100% ethanol for 1 hour. The tissue blocks were then blotted to remove absolute alcohol from their surface, after which they were carefully mounted on a tissue holder by applying 2 drops of Fevi kwik on to a wooden block. Sections were cut using a base sledge microtome. Coronal and horizontal sections of the hippocampus and coronal sections of the amygdala and motor cortex were cut at a thickness of 120 μm. The sections taken were further processed using a soft painting brush in the following order: Sections were collected in 70% ethanol, washed in distilled water for 5 minutes, 5% sodium carbonate for 20 minutes, distilled water for 5 minutes, 70% ethanol 10 minutes consisting of 2 washes for 5 minutes each, 90% ethanol for 10 minutes consisting of 2 washes for 5 minutes each, 100% ethanol for 10 minutes consisting of 2 washes for 5 minutes each, cedar wood oil for 1 hour, xylene for 10 minutes consisting of 2 washes for 5 minutes each. Sections were mounted on a glass slide using DPX. Clearing was presumed to be complete once the floating sections in the xylene started sinking. These sections were observed for their translucency. Translucent sections were mounted serially on a slide using DPX as the mounting media. After the cover slip had been placed, it was “banked up” with excess DPX on all-four sides. Care was taken to avoid inclusion of air bubbles. The slides were air dried horizontally for 1 week. Two days after mounting, ringing was done using DPX to prevent the entry of air bubbles into the slide.
Dendritic quantification
The dendritic quantification of hippocampal CA1, CA3, basolateral amygdala, and motor cortex neurons was done using the camera lucida technique. From each rat, 8–10 well-stained hippocampal CA1 and CA3 neurons and 8–10 basolateral amygdala and motor cortex neurons were traced using a camera lucida tracing device (Dutta Scientific Works, Bangalore, India). Only pyramidal neurons confined to the CA1 and CA3 regions of hippocampus were selected for tracing. All types of neurons from the different nuclei of amygdala and motor cortex were selected for tracing. Neurons that were darkly-stained throughout their arborization were selected. Neurons with truncated dendritic branches within a 100 μm radius from the cell body were excluded. Only those neurons that were relatively isolated from neighboring impregnated neurons and neuroglial cells were selected; based on our experience that densely-impregnated cells very close to each other interfere with the analysis. The concentric circle method of Sholl16 was used for dendritic quantification. On a transparent sheet, concentric circles were drawn. The radial distance between two adjacent concentric circles was equivalent to 20 μm. During dendritic quantification, the sheet with concentric circles was placed on the camera lucida-traced neuron so that the approximate center of the cell body of the neuron coincided with the center of the concentric circles. The number of branch points between two successive concentric circles i.e., within each successive 20 μm radial spheres were counted. The dendritic intersection was defined as the point where a dendrite touches or intersects the concentric circle. The number of dendritic intersections at each concentric circle was counted by placing the transparent sheet with concentric circles on the camera lucida-traced neurons. Both branch points and intersections were counted to a radial distance of 100 μm from the center of the soma.
Behavioral tests
Animals from each group were subjected to the following behavioral testing: T-maze test of spatial learning and the passive-avoidance test of short-term memory were performed on the 42nd day following group assignment for the normal controls, on the 42nd day following sham surgery for the sham-operated controls, on the 42nd day following lesioning for the KA-lesioned animals and on the 42nd day following lesioning for the KA-lesioned animals subjected to the immediate EE treatment (1-day after lesioning). T-maze and passive-avoidance tests were performed again on the 72nd day for the KA-lesioned animals subjected to the delayed EE treatment (60-d after lesioning). All testing was done at 7 pm, in accordance with the known diurnal variation of night-time activity in rats.
T-maze test
Animals were subjected to left-right discrimination, a spatial memory task. This task is a test of ability to discriminate between the left and right arm of a T-maze in order to acquire a food reward. Two days prior to the start of the testing, the rats were deprived of food to enhance motivation for the food reward. Subsequently, the food was restricted during trials so that the animal’s body weight was maintained at 85% of pre-test weight. Animals underwent a period of orientation during which time they were subjected to food restriction and were placed at the starting box for 60s. Rats were permitted to explore the T-maze for 30 minutes and to eat 15 pellets (10 mg each) of food in each goal area. After 30 minutes, the rats were returned to the starting box. This orientation procedure was carried out for 2 consecutive days. After the orientation period, six trials were conducted daily for four consecutive days.
Spontaneous alternation test
In each trial, the rat was first placed in the start box, and the rat was permitted to enter into the stem of the maze and choose either of the maze arms. A rat was considered to have entered into a particular arm only when it entered that arm with all its limbs. Once the rat ate a pellet in the goal area of that arm, the animal was placed back in the start box for the next trial. The inter-trial interval was 1 minute in duration. For each trial, the arm chosen by the rat was recorded. At the end of the 4-day experimental period (24 trials), the total number of alternations was recorded. The percent bias was computed for each rat using the following formula: percent bias = total number of selections of most frequently selected side / total number of trials × 100.
Rewarded alternation test
This test was initiated 1 day after completion of the spontaneous alternation test. During this test, 6 trials/day were conducted for 4 consecutive days. Each trial had 2 runs, a forced and a selection run. In the forced run, the animal was forced into one of the arms by blocking the other arm and was allowed to consume the pellets in the goal area. Once the animal ate the pellets in the goal area, it was placed back in the start box for a selection run. In the selection run, the goal area of the forced arm was kept empty and pellets were placed in the goal area of the opposite arm. Both arms were accessible for the rat; a 1 minute pause separated each forced and selection run, and a 1 minute pause separated each trial. The sequence of the forced arm was predetermined and was the same for all rats on a given day; on subsequent days, the sequence was alternatively changed. During the selection run, if the rat entered the arm opposite the forced arm, that response was recorded as “correct response.” If the rat selected the same arm that it had been forced to enter during the forced run, it was recorded as the “wrong response.” The percentage of correct responses was computed for each rat using the following formula: percentage of correct responses = total number of correct responses / total number of trials × 100.
Passive-avoidance learning and memory task
This task consisted of the following three parts: 1) an exploration test, 2) an aversive stimulation and learning phase (passive-avoidance acquisition), and 3) a retention test.
Exploration test
Each rat was subjected to 3 exploration tests on the same day. The inter-trial interval was 5 minutes and the duration of each trial was 3 minutes. Each rat was kept in the center of a larger compartment facing away from the entrance to a dark, smaller compartment. The door between the two compartments was kept open. The rat was allowed to explore the apparatus (both larger and smaller compartments) for 3 minutes. For each trial, the total time spent in the larger compartment, the total time spent in the smaller compartment and the number of crossings from the larger to the smaller compartment, a measure of exploratory behavior, were recorded. At the end of each trial, the rat was returned to the home cage, where it remained during an inter-trial interval of 5 minutes. This sequence was repeated three times for each rat.
Aversive stimulation and learning phase: passive-avoidance acquisition
After the last exploration trial, each rat was forced into the smaller compartment and the sliding door between the two compartments was closed. Three strong foot shocks (50 Hz, 1.5 mA, 1s duration) were given at approximately 5s intervals. The top cover was then opened and the rat was returned to its home cage.
Retention test
The retention test was performed 24 hours after the acquisition test. The rats were kept in the center of the larger compartment facing away from the entrance to the smaller compartment, the sliding door between the two compartments was kept open and each rat was allowed to explore the apparatus for 3 minutes; after 3 minutes, each rat was returned to its home cage. This sequence was repeated three times with an inter-trial interval of 5 minutes. For each trial, the total time spent in the larger compartment, the total time spent in the smaller compartment and the number of crossings from the larger to the smaller compartment, a measure of exploratory behavior, were recorded.
Results
Morphometric measurements in hippocampus, amygdala and motor cortex
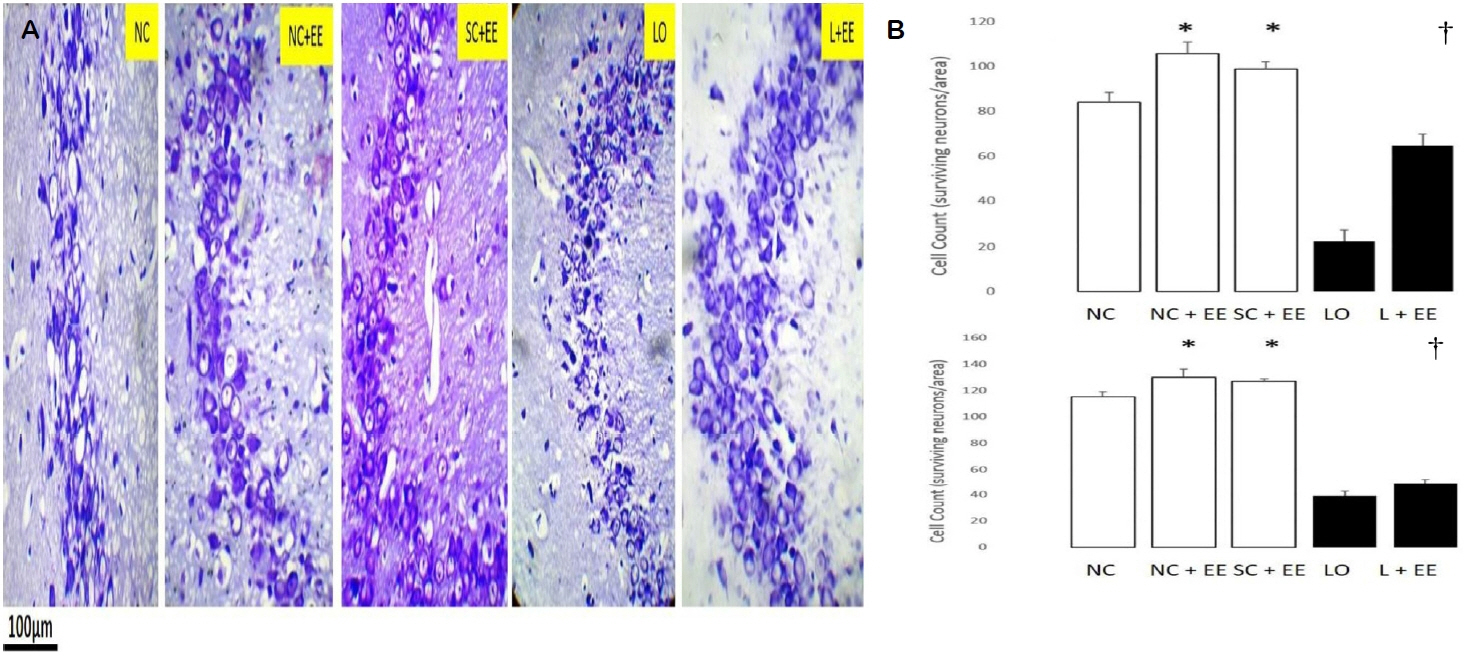
Fig. 2A shows the effects of EE, with and without KA lesioning, in the CA3 area of hippocampus through light microscopy using Nissl staining. Fig. 2B graphically depicts the effects of EE on the morphometric cell counts of surviving neurons in the CA3 areas of hippocampus. EE was associated with significant increases in the number of surviving neurons in the presence and in the absence of KA lesioning. These increases in surviving neurons were found in the animals subjected to EE 1 day and 60 days after grouping in the control groups and 1 day and 60 days after KA lesioning in the experimental groups.
Table 1 compares the counts of surviving neurons for the early EE (1 day postictal) and delayed EE (60 days postictal) in the CA3 region. The delayed EE intervention was associated with significantly fewer surviving neurons (68.5 ± 3.2 vs. 50.0 ± 1.6, p < 0.0001).
The effects of EE, with and without KA lesioning, on the surviving neurons in area CA1 and dentate gyrus, basolateral amygdala and motor cortex exhibited the same pattern as observed in hippocampal CA3 area (data not shown). EE produced a significant increase in the numbers of surviving neurons in these areas, both in the presence and in the absence of KA lesioning. Increased numbers of surviving neurons were also observed in the animals subjected to EE, 1 day and 60 days after grouping in the control groups and 1 day and 60 days after KA lesioning in the experimental groups, 60 days after grouping in the controls and 60 days after KA lesioning in the experimental group for area CA1, dentate gyrus, basolateral amygdala and motor cortex. Similar to the CA3 region, the number of surviving neurons was found to be significantly fewer in the groups subjected to delayed EE (60 days postictal) compared to those subjected to immediate EE (1 day postictal, data not shown).
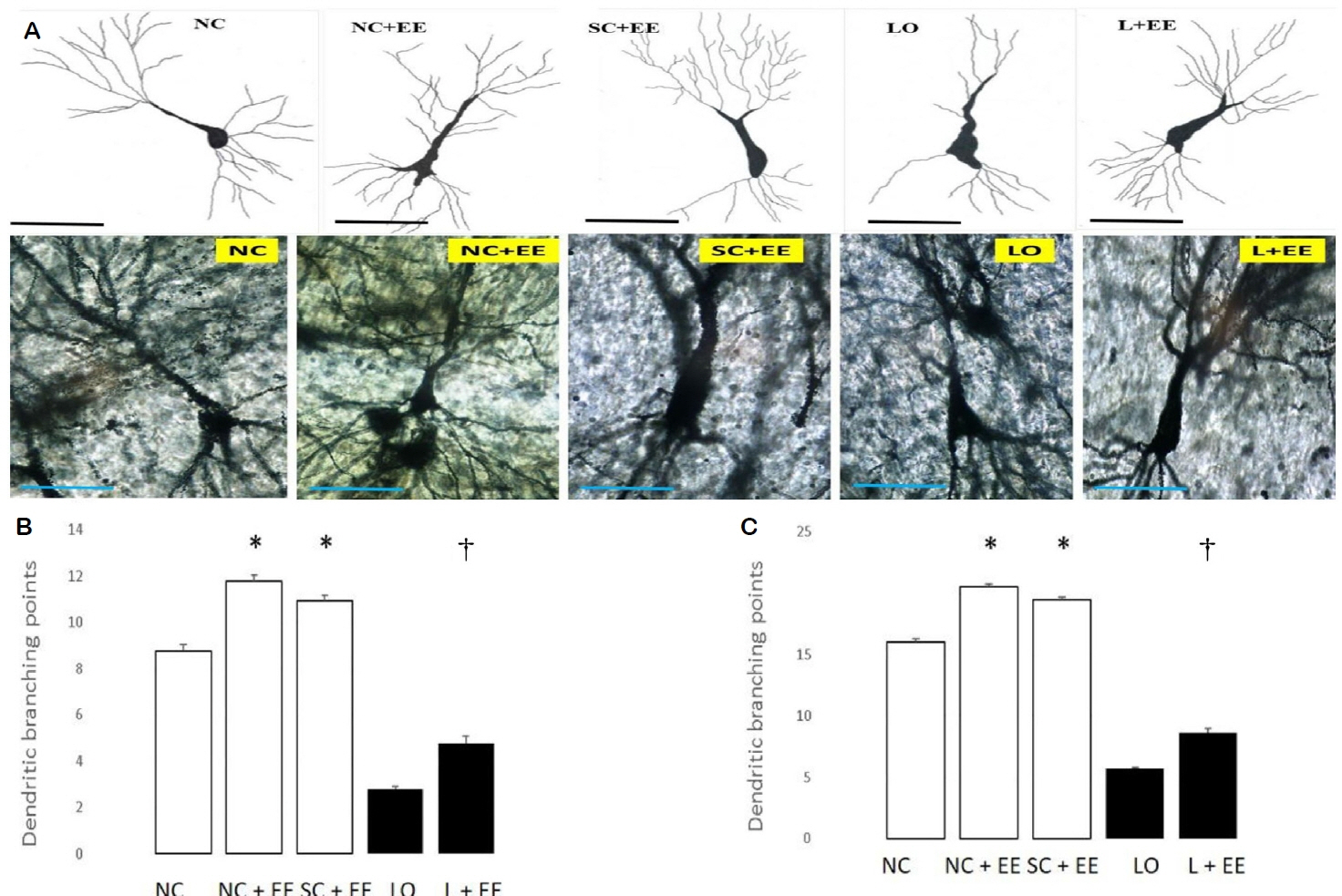
Fig. 3A shows the effects of EE on dendritic branching and intersection points of the surviving neurons in the CA3 area of hippocampus observed by Golgi-Cox staining and light microscopic camera lucida tracings. Fig. 3B, C depicts the effects of EE on the morphometric counts of branch points in the CA3 area of hippocampus. These data demonstrate that exposure to EE produced significant increases in the branch and intersection points with and without KA lesioning. The increases in branch points and/or intersections of surviving neurons were observed in the animals subjected to EE 1 day and 60 days after grouping in the control animals and 1 day and 60 days after KA lesioning in the experimental animals. The effects of EE on dendritic branch points and intersections of the surviving neurons in the CA1 area of the hippocampus, amygdala and motor cortex also demonstrated that EE increased the dendritic arborization with and without KA lesioning (data not shown). These increased numbers of branch points and intersections of the surviving neurons were seen in animals subjected to EE 1 day and 60 days after grouping in the control groups, as well as 1 day and 60 days after KA lesioning in the experimental groups for the following regions: CA1, dentate, amygdala and motor cortex.
Behavioral measurements
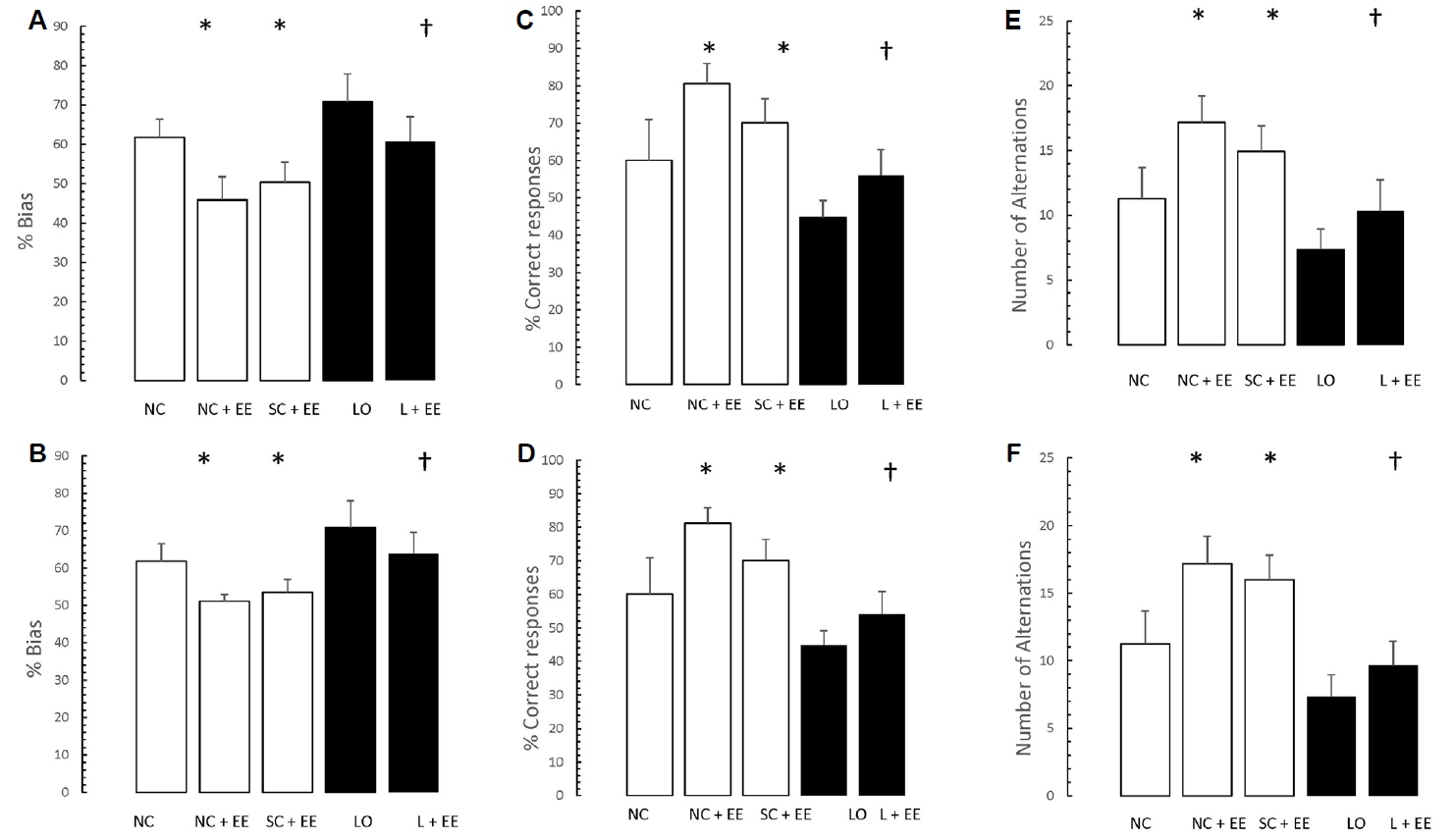
Fig. 4 depicts the effects of EE, with and without KA lesioning, on the results of T-maze testing. EE was associated with significant increases in the percent bias, percentage of correct responses and the number of alternations. Similar to the morphometric findings, these increases were found in the animals subjected to EE 1 day and 60 days after grouping in the control animals and 1 day and 60 days after KA lesioning in the experimental animals.
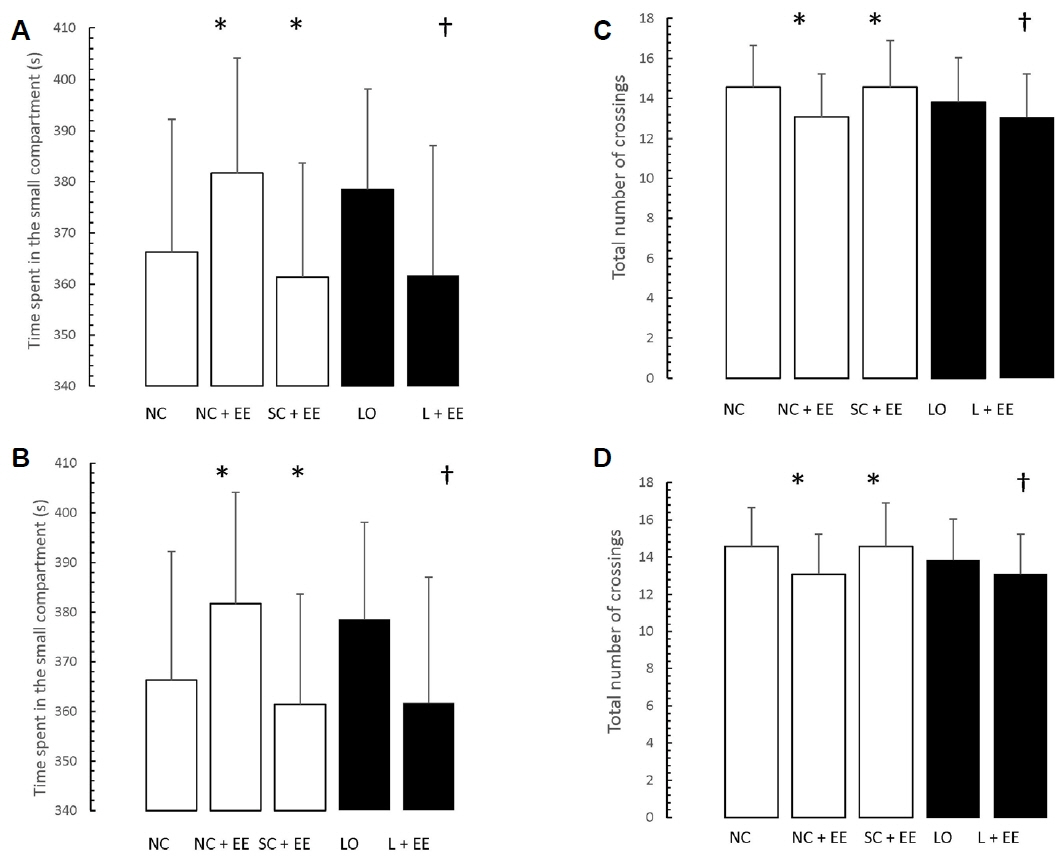
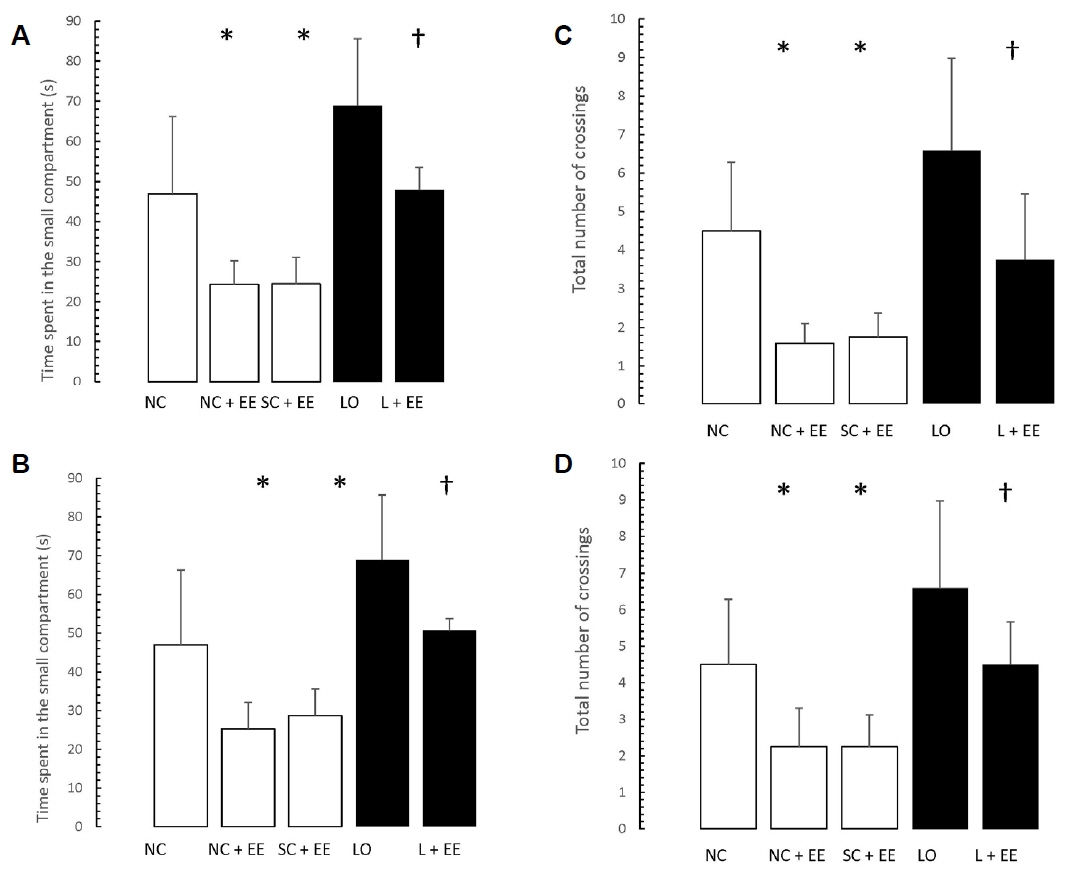
Figs. 5 and 6 present the effects of EE on the exploration and retention phases of passive-avoidance testing, respectively. EE was associated with no significant changes in the time spent within the smaller compartment and in the number of crossings, in the animals subjected to EE (Fig. 5), as well as significant decreases in the time spent within the smaller compartment where an aversive stimulus was previously administered as well as in the number of crossings (Fig. 6). Similar to the morphometric and T-maze data, these changes were observed in the animals subjected to EE 1 day and 60 days after grouping in the control groups, as well as 1 day and 60 days after KA lesioning in the experimental groups.
Table 1 shows that early vs. delayed exposure to the EE intervention resulted in no significant differences in either time spent in the small compartment where the aversive stimulus was given (memory test) or percentage of correct responses for the T-maze (learning test).
Discussion
The main finding of this study is that EE increased brain neurogenesis following chemically-induced (kainate) seizures, in hippocampus, amygdala and motor cortex. In conjunction with the morphological evidence of neurogenesis in these areas, EE also attenuated the learning and memory deficits associated with the kainate-induced seizures and augmented learning and memory. These morphometric and behavioral results were observed whether the EE intervention was initiated 1 day or 60 days, postictal.
One of the more interesting findings of this study is that, compared to the early exposure EE intervention, the delayed exposure was associated with significantly fewer surviving neurons in all the brain areas studied. However, we found no significant differences in dendritic arborizations related to the onset of EE exposure. This finding is easily explained by the fact that whereas the number of surviving neurons was expressed as a total for a given area, the dendritic branch points and intersections were expressed as numbers per surviving neuron. These findings suggest that the fewer surviving neurons associated with the delayed EE intervention expressed essentially the same cytoarchitecture as those associated with the early EE intervention.
Our control data demonstrated that identical EE interventions increased both neurogenesis and learning/memory functions in normal and sham-operated control animals, in the absence of seizures.
The effects of EE on neurogenesis in the hippocampal regions of CA1, CA3, and dentate gyrus basolateral nucleus of amygdala and motor cortex were demonstrated in the same animals. Increased neurogenesis was evidenced by increases in counts of surviving neurons and of their dendritic branch points and intersections. EE also increased the total number of neurons in all of the five brain areas studied in both the normal and the sham-operated controls. Physical exercise is known to be an effective stimulus for neurogenesis by virtue of positive correlations with cognitive functions in experimental animals, as well as in humans.8 Previous studies on rat brain neurogenesis show increases in the neural progenitor (stem) cells within the subgranular zone of dentate gyrus in the hippocampus and sub-ventricular zone in the olfactory bulb.17 However, neurogenesis is also seen in the CA1 and CA3 regions of the hippocampus, the amygdala3 and in the sensorimotor cortex18 but motor cortex neurogenesis has not been extensively studied.
Neurogenesis and brain signaling molecules
Nerve growth factor (NGF) is thought to protect against brain excitotoxicity.19 NGF undergoes retrograde transport and stimulates differentiation of progenitor cells.20 NGF is also a ligand for tropomyosin receptor kinase B (TrkB) and for p75 low-affinity nerve growth factor receptor (LNGFR)21 and in conjunction with brain-derived neurotrophic factor (BDNF), it stimulates neural stem cells.22–24 TrkB is thought to regulate neuroplasticity by protecting neurons from oxidative stress, thereby promoting regeneration.25 Simvastatin, niacin and low-level laser therapies are reported to upregulate both TrkB and BDNF and increase axonal and neurite growth in rats subjected to ischemia.26–28 BDNF is thought to promote neuroplasticity by stimulating dendrite growth and reorganizing synapses by production of postsynaptic density protein-95.29
EE and brain signaling molecules
Upregulation of neurotrophins has been widely reported following aerobic exercise in rats, and this is correlated with improved brain functions.27 Aerobic exercise has also been shown to be as effective as antidepressants and antihypertensive medications.27 Mice lacking BDNF exhibit depression-like behaviors.30 Similar to the effects of aerobic exercise, depression-like behavior in these mice is reported to be reversed more effectively by EE than by antidepressants.30 EE is also reported to upregulate the expression of neurotrophins in conjunction with increased expression of the NMDA receptors and of cAMP response element-binding in the hippocampus of both normal mice and mice genetically-modified to model Alzheimer’s disease.31 EE treatments of stressed (chronic immobilization) rats is shown to result in less behavioral depression/anxiety, spatial learning and memory impairment in conjunction with restoration of BDNF, vascular endothelial growth factor, glial fibrillary associated protein (GFAP) and glucocorticoid receptor expression, as well as, reversal of hippocampal atrophy.32 EE also reverses the decrements in neural precursor cell proliferation in brain neurogenic zones of the hippocampus and hypothalamus induced by genetic deletion of PPAR-alpha.33 It was beyond the scope of the present study to determine the molecular basis of the apparently beneficial effects of EE on seizures.
EE is reported to improve brain functions, including cognitive abilities in a mouse model of Down syndrome10 and in rat models of both Alzheimer’s disease and normal aging.11 Upregulation of neurotrophins has been widely reported following physical exercise in rats, and this has been correlated with improved brain functions.34,35 In previous studies from our laboratory, we reported that swimming induced improvements in neurogenesis and behavioral functions in the rat kainate model of temporal lobe epilepsy.36,37 Although we did not include a measure of physical activity in the present study, it is likely that our EE intervention was sufficiently stimulating to the animal subjects to increase their physical activity. Whether an EE intervention that does not stimulate physical activity could be an effective stimulus for neurogenesis in the kainate model of temporal lobe epilepsy remains an unanswered question. Future studies will, no doubt, address this question due to the likelihood that many humans suffering from temporal lobe epilepsy are, because of their disease or other factors, physically inactive. Such persons might benefit from behavioral interventions involving pure cognitive challenges such as reading, writing, problem-solving, academics, etc.
Experiment 1: EE, neurogenesis and learning in normal and sham-operated control rats
Our morphometric results of counting neuronal cell bodies, dendritic branch points and intersections showed similar patterns in each of the brain areas studied. Total numbers of neurons, dendritic branch points and intersections were increased significantly by EE whether the animals were exposed to the intervention immediately or after a delay of 60 days.
Our behavioral measurements were done in separate groups of control animals at two time points―immediately after grouping or sham operation, as well as 60 days after grouping or sham operation. It was beyond the scope of this study to determine the precise mechanism for the learning and memory improvements observed but, in accordance with previous research cited above, it is likely that the EE intervention increased the production of neurotrophic factors in the brain areas studied. In the normal control and sham-operated rats, percent bias was decreased, correct responses were increased, the number of alternations was increased on the T-maze task and the time spent in the small compartment was decreased, the number of crossings was decreased on the passive-avoidance task. These results were found after exposure to EE intervention whether EE was administered immediately or after a delay of 60 days following grouping or sham surgery.
Experiment 2: EE, neurogenesis and learning in kainate-treated rats
Kainate induced seizures in mice are reported to activate glia-like radial neural stem cells.38 These neural stem cells are thought to comprise a different subpopulation from those stimulated by running exercise.39 Glia-like radial neural stem cells are reported to migrate throughout the brain.40 The migration pattern of the neural stem cells stimulated by kainate-induced lesioning and seizures has previously been described.41 Our morphometric counting of cell bodies, dendritic branch points and intersections provides a measure of the number of neurons surviving kainate lesioning and the number of dendritic arborizations available for synaptogenesis.36 Our finding that EE increased the number of surviving neurons, dendritic branch points and intersections in all of the five brain regions and at the two time points studied suggests that there was a significant increase in neurogenesis whether the EE intervention was initiated and completed within the first 30 postictal days or after a delay of 60 postictal days. This finding is interesting because although EE appears to augment neurogenesis, persons affected by temporal lobe epilepsy may be unable to maintain the rigorous lifestyle required by EE, without breaks, over the long-term. Kainate-induced seizures in mice are reported to preferentially activate a specific subpopulation of cells, glia-like radial neural stem cells28,29 distinct from the subpopulation of neural stem cells stimulated by aerobic exercise.34 It is thought that such stem cells have the capacity to migrate throughout the brain.35 Our measurements of percentage of correct responses on the T-maze task and the time spent in the small compartment on the passive-avoidance task, as well as the other components of these tasks, support the hypothesis that the EE intervention may have increased the number of hippocampal neurons surviving the kainate lesioning and the number of dendritic arborizations available for synaptogenesis.36 The number of surviving neurons is reported to be correlated with the learning and short-term memory deficits observed in kainate-lesioned rats.8,9 These findings suggest that following kainate-lesioning and seizures, there probably was a significant increase in neurogenesis, mediating the learning and memory improvements, whether the EE intervention was initiated and completed within the first 30 postictal days or after a delay of 60 postictal days. This finding is significant because although EE may augment neurogenesis, humans incapacitated by temporal lobe epilepsy may be unable to maintain regular EE regimens, without breaks, over the long-term.
Limitations of the study
Identifying the neural stem cells and differentiating them from mature neurons would have provided a more direct measure of neurogenesis. We employed cresyl violet staining that labeled the Nissl substance and permitted counting of surviving neurons, dendritic branch points and intersections. We showed repopulation of the brain areas with mature neurons but could not count the number of neural stem cells present or their differentiation into neurons. Neuron-specific enolase,42 beta tubulin and/or nestin43 as neuronal markers and GFAP as neural stem cell marker could have more directly measured the process of neurogenesis. These immunohistochemical markers would have been more sensitive indicators of recovery than the counting of the surviving neurons marked with cresyl violet staining. We also did not do morphometric counting of surviving neurons, dendritic branch points and intersections in the brains of kainate-lesioned animals that were observed postictally, in the absence of the EE intervention. This experiment would have provided a measure of the effectiveness of spontaneous neurogenesis. Seizures alone have been reported to stimulate neurogenesis. More specifically, kainate-induced seizures are reported to be an effective stimulus for neurogenesis. An excitotoxic “limbic syndrome” is reported to occur at the sites of neurogenesis where damaged neurons continue to release substances which contribute to kindling of epileptic discharges. The presence of mossy fibers expressing kainate receptors is reported to result in the development of abnormal glutamatergic synapses.44–46 This process is thought to contribute to a vicious cycle of seizures often observed in patients affected by temporal lobe epilepsy. We observed that when individual kainate-lesioned rats were not housed in separate cages; i.e., when they were housed communally, they had a tendency to cannibalize each other. This behavior may be a model for the emotional dysregulation reported in temporal lobe epilepsy patients who are resistant to treatment with antiepileptic drugs.47
The main limitation of the behavioral learning and memory component of this study is that we employed manual methods for recording our data and did not have access to computer-based learning and memory tasks. A radial rather than a T-maze apparatus and a water-maze, rather than a two-compartmental, apparatus for passive-avoidance testing would have permitted automated, electronic recording of the data. These methods would likely have increased the sensitivity of our behavioral measurements, likely demonstrating greater deficits and improvements in learning and memory in the kainate-lesioned rats, perhaps with fewer trials and animals. The numbers of trials and animals we employed was more than adequate, as shown by the levels of statistical significance, and the study had sufficient statistical power. The aforementioned computer-based methods might have made our results more robust, but is unlikely to have changed the interpretation.
The main results of this study demonstrate that regular exposure to a regimen of EE, following kainate-induced seizures in rats, increases the number of surviving neurons, dendritic branch points and intersections in centers for emotional regulation and motor coordination. Considering, neuronal regeneration via neurogenesis is the basis of neural plasticity, these findings seem to support the hypothesis that EE may improve the clinical outcomes of temporal lobe epilepsy patients by the mechanism of increasing neuroplasticity. Future studies should evaluate the effects of EE involving various forms of pure cognitive and academic challenges, in the absence of the confounding factor of increased physical activity, in temporal lobe epilepsy patients.
The results of this study also demonstrate that EE has the capacity to attenuate the learning and memory deficits in rats subjected to kainate-induced seizures, whether there is a 60-day delay in initiating the exercise treatment. Learning and memory deficits in the rats subjected to the seizures immediately, without the 60-day delay, were found to be less than those in the animals that were exposed to EE after a 60-day delay. Previous studies demonstrate correlation between learning/memory deficits and neuronal loss in the hippocampus was associated with kainate-lesioning and temporal lobe seizures. These findings imply that EE may decrease learning and memory losses even after a substantial delay following temporal lobe seizures. Patients diagnosed with temporal lobe epilepsy and other seizure disorders are often debilitated, depressed and lack the motivation to study, work or maintain effective interpersonal relationships;48,49 thereby, constituting a form of environmental impoverishment.50 Future studies should take into consideration that the current clinical guidelines on medical management of temporal lobe epilepsy do not include EE as an adjunct to antiepileptic drug treatments.