Juvenile myoclonic epilepsy (JME) is an idiopathic, age-related generalized epileptic syndrome, in which the onset age of seizures coincides with adolescence, i.e. from 12 to 18 years, and a peak at age 15. Absence seizures are often the first type, occurring in 40% of the cases, followed by generalized tonic-clonic (GTC) and myoclonic seizures [1]. In the literature, there were some reports about paradoxical aggravation of idiopathic generalized epilepsy after treatment with antiepileptic drugs, such as carbamazepine and phenytoin [2]. However, except some case reports, status epilepticus (SE) in JME is very rare, and little is known about its etiology [3]. Most of the cases were in children or in adults who were already diagnosed as JME [4–6]. We report two previously healthy women, who were presented with first-ever status epilepticus.
Case Report
Patient 1
A 52-year-old woman presented with generalized tonic-clonic seizure (GTCS) at the first seizure attack. In general, she was healthy, and only suffered from mild headache in the past few months.
During the night before visiting the ER, she showed inappropriate behavior, confusion, and disorientation. A few hours later, she showed GTCS lasting for 1–2 minutes, and then, remained in a mild confusion state in the ER. There were no abnormal findings on the brain MRI and in the CSF analysis.
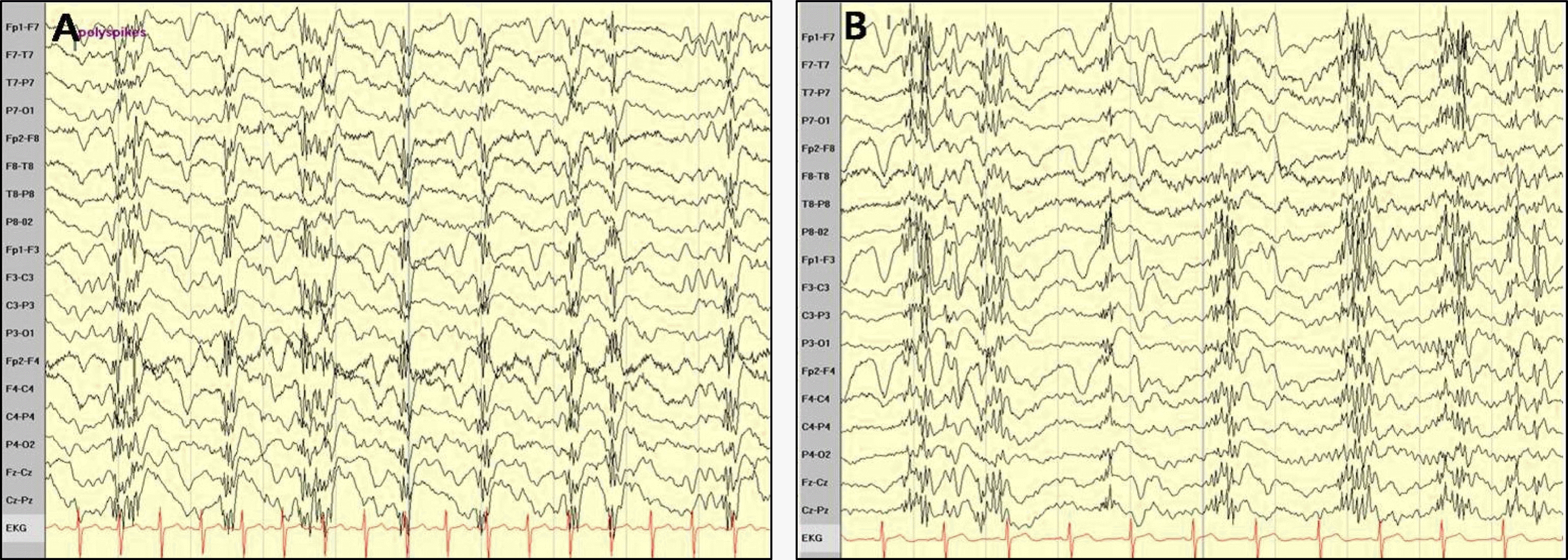
During the confusion state, on the EEG, there were repetitive, high voltage generalized polyspikes and waves with flat periods (Figure 1A) which subsided after intravenous injection of lorazepam. Detailed history taking revealed previous occasional absence-like episodes and myoclonic jerks. The patient’s younger sister also suffered from epilepsy since childhood.
The epileptic discharges were all abolished on the follow-up EEG.
The patient was diagnosed as JME and there has been no recurrence of seizures with sodium valproate medication.
Patient 2
A woman, aged 39 visited the ER with transient loss of consciousness (LOC). W Anamnesis showed that, when she was 27 years old, during excessive working at night, she experienced one episode of LOC without convulsions.
Both the brain MRI scan and the EEG performed at that time were normal. Recently, she felt exhausted because of her night job. In the ER, she was alert and could follow all commands, but showed mild confusion. Focal neurological signs were not observed, and brain imaging was normal. Generalized polyspikes and slow waves repetitively occurred on the EEG (Figure 1B).
The pathologic waves disappeared after IV injection of lorazepam.
The EEG recorded on the following day showed normal background activity and decreased slow wave activity.
With careful history taking, we found out that she also experienced a myoclonic jerk soon after awakening. She was diagnosed as JME, and no further seizures occurred after sodium valproate administration.
Due to the occurrence of hand tremor, the sodium valproate treatment was changed to lamotrigine during the follow-up period.
Discussion
We report 2 cases diagnosed as JME for the first time in their adult life. Diagnosis was expressed by a brief convulsive seizure in the first patient and by LOC following NCSE in the second patient.
Status epilepticus is uncommon in the idiopathic generalized epilepsy syndrome. Moreover, among the etiologies of NCSE, JME is very rare. Thomson et al. [6] studied 32 patients who were initially diagnosed as NCSE, and identified that only 1 of these patients had a history of juvenile myoclonic epilepsy and 5 of them had absence epilepsy. Rainer et al. [7] retrospectively reviewed 69 patients with JME, and found that only 4 patients developed NCSE during the first 6 years of their follow-up period. Their findings, as well as the low prevalence of NCSE in JME (estimated at 5.8%) and the incidence of only 1.2 per year, suggest that NCSE in patients with JME is indeed rare.
Most of the patients with JME experience myoclonic jerks, absence, and/or generalized tonic-clonic seizures within the 2nd decade of their lives. Despite clear cut clinical symptoms and age distribution, the diagnosis of JME is often delayed. It is commonly considered as partial epilepsy, (especially in patients with no history of myoclonic jerks) rather than absence seizures or GTCS [8,9]. The most important factor for misdiagnosis is failure to detect the myoclonic jerks. Most patients do not complain of myoclonias at first, because either they find them to be unimportant or they are not perceived as abnormal [8]. In addition, clinicians frequently miss the history taking of myoclonias during the medical interview, a situation which was common for the majority of the investigated patients [9]. In our two presented cases, detailed history taking showed that the patients already had absence-like episodes and myoclonic jerks, but, similar to previous cases, they did not consider them as abnormal.
When approaching the adult patient with mental change, the EEG is essential to differentiate the NCSE from another cause of mental change. There are usually 3 forms of NCSE in patients with idiopathic generalized epilepsy patients: 1) typical absence SE, 2) atypical absence SE, and 3) complex partial absence SE. In theory, the EEG of the typical absence SE exhibits trains of 3 Hz spike-and-wave complexes which are easily differentiated from the EEG of both the atypical absence SE and the complex partial absence SE. However, the typical absence SE can show an irregular frequency (from 1–4 Hz) of the spike-wave or polyspike-wave complexes. Furthermore, the complex partial SE can also present with generalized spike-wave discharges. In such cases, the response to treatment can be helpful for orienting the diagnosis. Thus, in patients suffering from partial SE with generalized EEG activity treated with IV benzodiazepine, the focal features are unmasked by the treatment.
Furthermore, it has been shown that the postictal state associated with EEG slowing is more prolonged in the partial SE [3]. Our patients showed generalized, irregular polyspike-wave complexes without myoclonus. The abnormal EEG features of our patients was well controlled after intravenous injection of lorazepam, without any postictal focal abnormality.
The correct diagnosis and proper treatment is essential for these patients, considering that the narrow spectrum antiepileptic drugs, such as phenytoin, carbamazepine, gabapentin, which are commonly used in adult onset seizures, may worsen the seizures or even cause SE in JME [9,10].
Status epilepticus in patients with JME is rare but we could learn through our cases that it can be seen sometimes in the clinical setting. Moreover, in the adult patient with first ever status epilepticus (i.e. without obvious history of epilepsy), the clinicians can often neglect the possibility of primary generalized epilepsy and can therefore miss the correct diagnosis. Detailed history taking of either myoclonus or absence like episodes, the EEG features, and the response to treatment may be helpful for the differential diagnosis.
In conclusion, ecognition and suspicion of the disorder and appropriate management is mandatory.