Introduction
Epilepsy affects 1–2% of children and 0.5–1% of the population worldwide. Approximately 30% of patients with epilepsy have refractory epilepsy, that is, seizures persist despite accurate diagnosis and carefully monitored treatment with antiepileptic drugs.1,2
Kindling is a model of epilepsy, with the advantage of being both an epileptogenic and a spontaneous seizure model. Kindled seizures are widely accepted as an animal model of human temporal lobe epilepsy (TLE), wherein repeated sub threshold brain stimulation (electrical or chemical) leads to behavioral signs of seizures (tonic and clonic)3,4 and such procedure results in a state mimicking many signs of human TLE: hippocampal atrophy, loss of neurons in the limbic areas: Cal, CA3, and dentate gyrus of the hippocampus, glass and growth of new neuronal connections.5–7
Studies have focused on the role of oxidative stress in seizures. Free radicals have been implicated in the development of seizures.8,9 Animal data also suggest that even a single seizure may be damaging and can cause DNA fragmentation which is the characteristic feature of apoptosis. Also Gamma-enolase, a neuron-specific enzyme critical in energy metabolism, is elevated following anoxia, stroke, or status epilepticus.10
Peroxisome proliferator-activated receptors (PPARs) have been considered as potential drug targets for seizure control.11 Both PPAR α and PPARγ are activated by the polyunsaturated fatty acids (PUFA) and their eicosanoid derivatives, as well as by synthetic legands such as the fibrates.12 Activation of peroxisome proliferator-activated receptors stimulates the transcription of the genes involved in fatty acid oxidation via the formation of heterodimeric transcription-factor complexes with the retinoid-X-receptor (RXR).13
Based on animal studies that have shown that selective agonists of PPAR α or PPAR γ raise seizure thresholds, it is suggested that PPARs may be involved in seizure control.14 Fenofibrate, for instance, an agonist of PPAR α, has been reported to raise the seizure threshold in the acute seizure models like tail-infusion pentylenetetrazole (PTZ) and Pilocarpine seizure tests in rats.15 The involvement of of PPARα in seizure control has been documented by Puligheddu M et al in nicotine induced seizures in mice and rats.16 The role of PPARα in a genetic model of absence epilepsy has also been demonstrated in a study by Citraro R et al in rat.17 However, the role of PPAR-α in the kindling seizure models have not been reported in the published literatures.
The purpose of the present study was to evaluate the anti kindling effects and the mechanism of bezafibrate, a PPARα agonist in PTZ induced kindling model of seizures in rats. These findings may provide insights into the understanding of the mechanism of bezafibrate as an anti kindling agent and could offer a useful support to the basic antiepileptic therapy in preventing the development of PTZ induced seizures, suggesting its potential for therapeutic applications in temporal lobe epilepsy.
Methods
Animals
Young male wistar rats (200 to 250 gm) were housed 2 per cage and maintained at 23±20 C with a relative humidity of 65±5% in 12 hours light/dark cycle. Animals were acclimatized to laboratory conditions for at least 7 days prior to experimentation. All procedures involving animals and their care were performed with the agreement of the local ethics committee for animal experimentation and in compliance with our institutional guidelines, which in turn comply with current national and international laws and recommendations.
Drug preparation and dosing schedule
PTZ was dissolved in 0.9% saline in strength of 0.31% and injected intraperitoneally (i.p.) in a volume not exceeding 1.0 mL/kg, at a sub convulsive dose of 30 mg/kg every alternate day until development of kindling or up to 10 weeks. Bezafibrate (100, 200, 300 mg/kg) was dissolved in 5% dimethyl sulfoxide (DMSO) and Sodium Valporate (200 mg/kg) in 0.9% saline and were given intraperitoneally, 30 minutes before the PTZ injection till the animal develops kindling or up to 10 weeks. All chemicals were procured from Sigma Pharmaceutical Industry Co.
Diets and treatments
Animals had free access to standard pellet chow diet and tap water ad libitum. Rats were weighed and divided into 7 groups: Group I: Vehicle Control (DMSO 5%+PTZ 30 mg/kg) (n=8); Group II: Saline Control (Normal Saline+PTZ 30 mg/kg) (n=8); Group III: sodium valporate (VPA) 200 group (Sodium Valproate 200 mg/kg+PTZ 30 mg/kg) (n=8); Group IV: bezafibrate (BEZF) 100 group (Bezafibrate 100 mg/kg+PTZ 30 mg/kg) (n=8); Group V: BEZF 200 group (Bezafibrate 200 mg/kg+PTZ 30 mg/kg) (n=8); Group VI: BEZF 300 group (Bezafibrate 300 mg/kg+PTZ 30 mg/kg) (n=8), Group VII: unanaesthetized group. To see the impact of pentobarbital on apoptotic biomarkers and oxidative stressed parameters, an unanaesthetized group was included where animals were sacrificed by stunning.
Pentylenetetrazole induced kindling in rats
PTZ was injected i.p. as mentioned above. After each injection of PTZ, the rats were placed singly in isolated transparent Plexiglas cages and were observed for 2hrs/day. The intensity of convulsions was rated according to modified Racine scale as follows:18 0-No response; 1 - Ear and facial twitching; 2 - Myoclonic jerks without rearing; 3 - Myoclonic jerks with rearing; 4 - Turn over into side position, clonic - tonic seizures; 5 - Turn over into back position, generalized tonic - clonic convulsions. An animal was considered kindled when it exhibits, stage 4 or 5 of seizure score on three consecutive trials.
Collection of samples
When the animal became fully kindled (exhibits, stage 4 or 5 of seizure score on three consecutive trials), on the next day, it was sacrificed by decapitation under the overdose of intraperitoneal pentobarbitone (50 mg/kg) anesthesia. The hippocampus was carefully dissected out of the brain. 2 mL of blood was also collected from each anesthetized animal by cardiac puncture prior to decapitation.
Studies with hippocampus
Histopathology of the hippocampus
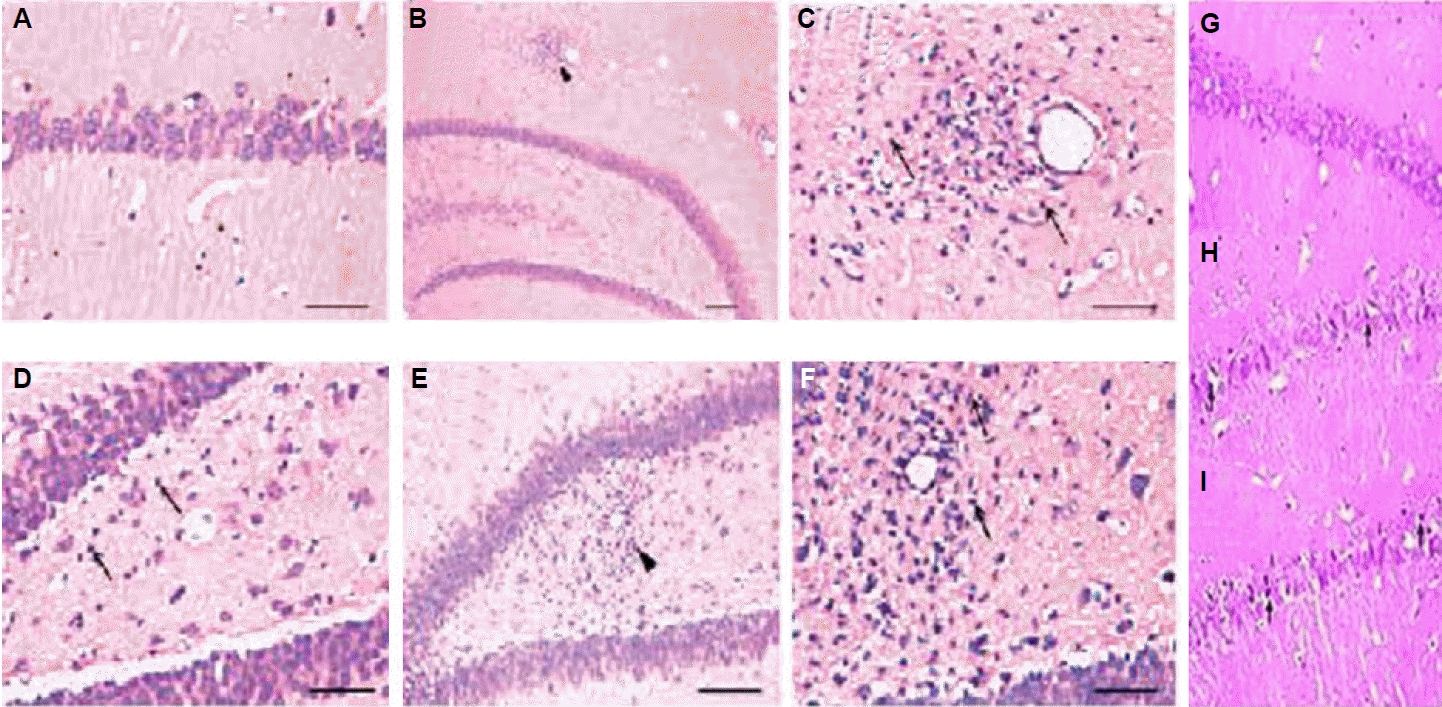
One half of each brain was fixed in 10% formalin and stored for histopathological studies using hemotoxylin and eosin (H&E) stain. Thereafter, tissue was sliced, routinely processed, and embedded in paraffin wax. 5 μm coronal paraffin sections were cut, mounted and stained by haematoxylin and eosin. The acidophilus neuron, identified by intense cytoplasmic eosinophilia accompanied by chromatin dispersion with loss of nuclear membrane integrity, was perceived as the marker for irreversible neuronal damage at the cellular level. The numbers of acidophilus neurons in different regions of the hippocampus were estimated on a 0–3 grading scale, 0=none, 0.5=slight (<10%), 1.0=mild (10–25%), 1.5=mild-to-moderate (26–45%), 2.0 =moderate (46–54%), 2.5=moderate-to-severe (55–75%), and 3.0 =severe (>75%), as previously published.19 The surviving cells were de ned as round-shaped, cytoplasmic membrane-intact cells, without any nuclear condensation or distorted aspect. The surviving pyramidal cells in the hippocampal CA3 region were seen at high magnification (400×). Sections were examined by a blinded investigator without knowledge of any other data on that animal.
Hippocampal lipid peroxidation studies
Estimations of thiobarbituric acid-reactive substance
The extent of lipid peroxidation was estimated according to the method of Ohokawa et al20 in tissue homogenate. Tissue homogenate was prepared in a ratio of 1g of wet tissue to 9 mL of phosphate buffer (pH 7.2) using a homogenizer. To 0.1 mL of the homogenate, 0.2 mL of 8.1% sodium dodecyl sulfate, 1.5 mL of 20% acetic acid solution, 1.5 mL of a 0.8% aqueous solution of thiobarbituric acid were added. The mixture was finally made up to 4.0 mL with distilled water, and heated at 95°C for 60 min. After cooling with tap water, 1.0 mL of distilled water and 5.0 mL of the mixture of n-butanol and pyridine (15:1, v/v) were added, and the mixture was shaken vigorously. After centrifugation at 4,000 rpm for 10 min, the absorbance of the organic layer (upper layer) was measured at 532 nm in a spectrophotometer against a blank containing all the reagents except the homogenate. The malondialdehyde (MDA) equivalents of the samples were calculated using the extinction coefficient 1.56×105 M−1cm−1.
Determination of catalase activity
The activity of catalase was measured by the method of Luck.21 A 10% w/v homogenate of the hippocampus was prepared in phosphate buffer. The homogenate was centrifuged and the supernatant was used for the enzyme assay. In short, the reaction mixture contained Tris (50 mM) - EDTA (5 mM) buffer, pH 7.0, 10 mM H2O2 (in 0.1 M KH2PO4 buffer, pH 7.0) in test cuvette. The reference cuvette contained Tris-EDTA solution and distilled water only. The contents of both the cuvettes were incubated at 37°C for 10 min. The reaction was started by the addition of tissue homogenate to both reference as well as test cuvettes. The rate of elimination of H2O2 by catalase was measured by recording the rate of change of absorbance per min at 240 nm for 4 min. Catalase activity was expressed as μmol of H2O2 consumed/min/mg protein using a molar extinction coefficient of 43.6 mM−1cm−1.
Reduced glutathione estimations
Assay of glutathione (GSH) was performed in tissue homogenates by the method of Moron et al.22 To 500 μL homogenate 100 μL of 25% trichloroacetic acid (TCA) was added. The precipitated proteins were separated by centrifugation at 2,000×g for 15 min. Supernatants were diluted with 0.2 M sodium phosphate buffer, pH 8.0. To this, 2.0 mL of 0.6 mM 5,5 Dithiobis (2-Nitrobenzoic acid) was added. The colored complex formed by DTNB and GSH was measured spectrophotometricaly at 412 nm against a reference cuvette containing 0.1 or 0.2 mL of 5% TCA. A standard curve of GSH was plotted with every set of assays. All the assays were done in duplicates. The levels of GSH were expressed as μg of GSH/mg protein.
Measurement of serum neuron specific enolase
2 mL of blood was collected from each anesthetized animal by cardiac puncture prior to decapitation. Blood samples were allowed to clot for 2 hours at room temperature or overnight at 4°C before centrifugation for 15 minutes at 1,000×g. Serum was removed and samples were stored at −20°C or −80º. Repeated freeze-thaw cycles were avoided. The sample was subjected to the estimation of Neuron-Specific Enolase (NSE) by ELISA kit (CUSABIO BIOTECH CO., LTD., China) according to the manufacturer’s instructions.
Results
Effect of bezafibrate in PTZ-induced kindling model
In vehicle control and saline control groups, the rats showed a gradual increase in the seizure score throughout the 10 weeks of the study period and 7 animals out of 8 (87.5%) became kindled at the end of 10 weeks in both the groups (Table 1, Fig. 1).
Sodium valproate (200 mg/kg, ip) pre-treatment significantly reduced the seizure score in the PTZ treated animals throughout the 10 weeks of study period (Table 1). In the VPA 2,000 group, none of the animal developed kindling at any point of time. The data for sodium valproate treated group were significantly less as compared to that of vehicle control and saline control group at all time points (p<0.05, Fig. 1).
Bezafibrate showed dose-dependent protection against PTZ induced kindling in rats. At 200 mg/kg, the effect of bezafibrate was significantly better when compared to that of 100 mg/kg of bezafibrate. However, the beneficial anti kindling effect of bezafibrate reached a ceiling at 200 mg/kg dose, since the maximum seizure score was 1.96±0.15 in 200 mg/kg group as compared to the maximum seizure score of 2.58±0.30 in 300 mg/kg group which was statistically significant. The anti kindling effect (particularly the seizure score) of bezafibrate was significantly less as compared to that of sodium valproate. At 200 mg/kg, bezafibrate showed an anti kindling effect which increased over the period of study and was significantly better as compared to that of vehicle control and saline control groups from the 5th week onwards till the end of the study period i.e. 10th week both in terms of reduction in seizure score and the number of animals developing PTZ induced kindling behavior. In the bezafibrate groups, the seizure score increased gradually up to week 5 and then gradually decreased because chronic administration of bezafibrate takes some time to produce its effect on the seizure scores and also due to animals being withdrawn because they have met the full-kindled criteria. Whereas in the control groups, the seizure scores increase consistently, which indicate that in the control group individual animal shows high seizure scores. Sodium valproate on the other hand, showed a significant anti kindling effect from 1st week onwards till the end of the study (Table 1).
Lipid peroxidation parameters
Effect of bezafibrate on reduced glutathione levels in the rat brain
Treatment with PTZ in vehicle control and saline control groups significantly reduced the GSH levels in rat brain when compared with unanaesthetized normal control rat. Pre-treatment with sodium valproate (200 mg/kg) and three different doses of bezafibrate (100 mg/kg, 200 mg/kg, 300 mg/kg, i.p.) significantly increased the level of GSH in brain towards the normal. Among all the three doses of bezafibrate 200 mg/kg was more effective in increasing the GSH level in the brain as compared to that with 100 and 300 mg/kg doses (Table 2).
The effect of bezafibrate on malondialdehyde levels in rat brain
Pre-treatment with sodium valproate (200 mg/kg) and three different doses of bezafibrate (100 mg/kg, 200 mg/kg, 300 mg/kg, i.p) also significantly reduced the whole brain MDA level in PTZ treated animals and it was statistically significant when compared to vehicle and saline treated groups. The reduction in the brain MDA level was significantly more with bezafibrate 200 mg/kg pre treated group when compared to bezafibrate 100 and 300 mg/kg pre treated group (Table 2).
The effect of bezafibrate on catalase activity in rat brain
The catalase activity was significantly decreased in vehicle and saline treated rats when compared to the unanaesthetized control animals. In the sodium valproate and bezafibrate pre treated groups, the catalase activity in rat brain was comparable with that of the control group but significantly higher as compared to that of the vehicle and saline treated groups (Table 2).
Effect of bezafibrate on serum neuron specific enolase levels in rats
The level of NSE in serum was significantly elevated in vehicle and saline treated group as compared to the unanaesthetized control group. In the sodium valproate pre treated group (6.12±0.28 nmol/mg protein), there was significant reduction in serum neuron specific enolase (sNSE) level as compared to that observed with vehicle and saline treated groups. All the three doses of bezafibrate also showed significant reduction in sNSE level as compared to vehicle and saline treated groups. Bezafibrate at 200 mg/kg and 300 mg/kg showed significant reduction in sNSE levels as compared to that with bezafibrate 100 mg/kg, while the effect observed with the dosage of 200 mg/kg was most effective and was statistically significant as compared to that with 300 mg/kg dose of bezafibrate (Table 2).
Histopathological changes in the hippocampus
We rarely observed acidophilic neurons in different sectors of the PTZ-treated rats’ hippocampus. Despite lacking of evidence for PTZ-induced cell necrosis, our study found gliosis in area CA1 and the hilus of the hippocampus in PTZ treated rats (Table 3, Fig. 2), suggesting the existence of neuronal loss. In VPA 200 group, there was less or no gliosis. Pre-treatment of BEZF 200 and 300, there was also less gliosis as compared to PTZ group. H&E staining showed that dead neurons in hippocampal CA3 region with pyknotic nuclei are clearly distinguishable from surviving cells that show round and palely stained nuclei. As shown in Fig 3A and 3B, the surviving neuron numbers in PTZ alone treated group were signi cantly decreased compared with that of control group (p<0.05). Moreover, BEZF 200, signi cantly attenuated the neuron loss induced by seizures (p< 0.05) (Fig. 3A and 3B).
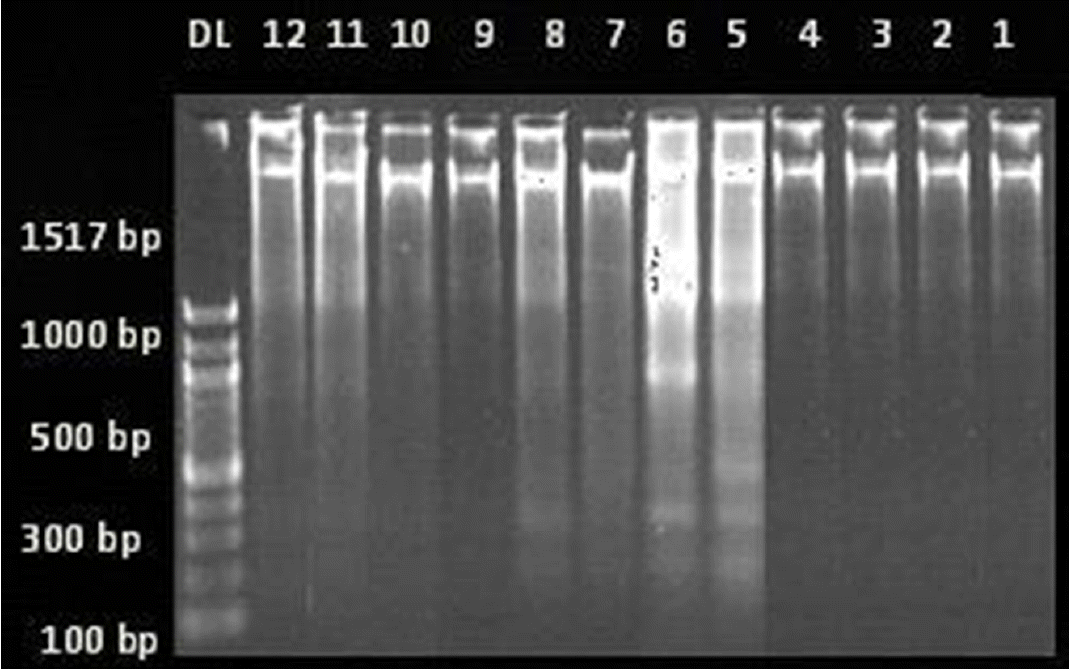
DNA fragmentation studies in the hippocampus of PTZ induced kindled rats
All the brain samples from different groups were subjected to DNA fragmentation. The PTZ and BEZF 100 groups showed laddering pattern with the formation of a large number of fragments ranging from 1,517 base pairs to 100 base pair fragments indicating DNA fragmentation. In contrast, VPA 200, BEZF 200 and BEZF 300 groups did not show formation of small fragments and hence no prominent laddering pattern (Fig. 4).
Discussion
In the present study, bezafibrate exhibited a marked non dose dependent anti kindling activity, suggesting its beneficial role in reducing seizure severity. Bezafibrate pre-treated groups showed significant reduction in the percent of animals (12.5%) developed kindling. PTZ thresholds were increased and the onset of seizure was delayed in the PTZ-induced kindling model.
As per a previous study, a PPAR-γ ligand (FMOC-Lieucine), exerted anticonvulsant properties against audiogenic seizures induced in adult magnesium-deficient mice.23
However, in the same model, rosiglitazone, other PPAR-γ agonist, did not protect mice. The seizure protection induced by FMOC-L-Leucine was also reduced by a PPAR-γ antagonist. Study on PPAR-α agonist, fenofibrate has reported the anticonvulsant property of the drug comparable to kitogenic diet in adult rats using PTZ induced acute seizure model and lithium -Pilocarpine induced status epilepticus (SE) model.15
PPAR-α is likely to regulate the expression of many genes that encode enzymes of fatty acid oxidation, ketogenesis, amino acids, and neurotransmitters in the brain and/or liver.24–27 One principal mechanism of action proposed for antiepileptic drugs is their ability to correct disturbances in neurotransmitter concentrations.28,29
Recent years have focused on the role of oxidative stress in seizures. Free radicals have been implicated in the development of seizures, causing cellular dysfunction by attacking at the polyunsaturated sites of the biological membranes leading to lipid peroxidation.8,9 Frantseva et al,8 using mice with amygdala convulsion, showed increased oxygen radical production and lipid peroxidation following a seizure, which was inhibited using antioxidants. A study by Gupta YK, et al,9 also demonstrated increased oxidative stress in PTZ induced kindling model in rats. Some antioxidants have been shown to be effective in reducing the oxidative stress in the models of epilepsy.30–32
In our study PTZ also produced oxidative stress and there was a significant increase in the brain MDA levels (an indicator of the lipid peroxidation due to free radical generation), reduction in GSH (endogenous antioxidant) levels and decreased catalase activity as compared to control. Both sodium valproate and bezafibrate reversed this increase in brain MDA levels and reduced glutathione levels in addition to elevating brain catalase activity which was found to be at par with the previous studies where bezafibrate showed anti-oxidative properties.33,34 Possibly, the antioxidant property and free radical scavenging potential of bezafibrate which preserved the antioxidant enzyme catalase activity, consequently lead to reductive effect on seizure intensity and duration.
Studies by Cavazos JE, et al35 and Pitkanen et al36 have shown that seizure activity is associated with neuronal damage and both necrotic and apoptotic forms of cell death contribute to brain damage in the PTZ-induced epilepsy model. Also, as per previous studies, chronic treatment with sodium valproate after kainate-induced SE prevented spontaneous recurrent seizures and reduced histological lesions in the hippocampus.37,38 Bezafibrate also proves to be a promising agent for the treatment of neurodegenerative diseases.39
In the present study, sodium valproate and bezafibrate maintained near normal morphology of the neurons while PTZ kindled rats showed diffuse neuronal injury with most of the neurons demonstrating nuclear chromatin clumping, hypereosinophilia, condensation of the cytoplasm and fragmentation of the cells.
Neuron specific enolase, a sensitive marker of neuronal damage increases in animal models of traumatic and ischemic brain injury, cerebral hypoxia, and epileptic seizures.40–45 Study by Wang et al,46 has demonstrated increased level of sNSE in PTZ kindled rats and pre treatment with topiramate and folic acid decreased sNSE levels.46 Our data also showed that there is an increase in sNSE in vehicle and saline treated groups and decrease in sodium valproate and bezafibrate pre treated groups, accompanied by histological evidence of neuronal damage in the hippocampus. Thus we postulate that the increased sNSE in vehicle and saline control groups may be a consequence of neuronal injuries in the hippocampus and bezafibrate decreased the sNSE by reduced the oxidative stress induced neuronal injuries in the hippocampus. In order to investigate the extent of neuronal cell damage caused by PTZ induced epileptic seizures, we carried out nuclear DNA fragmentation studies. In a study, Zhand et al demonstrated that in amygdala kindled seizures with increasing seizure severity, DNA fragmentation increases.47 In the present study all the brain samples of the PTZ treated group showed laddering pattern indicating DNA fragmentation. While the sodium valproate and bezafibrate groups did not show any major fragmentations.
These findings suggest, seizures cause an early production of oxidative damage to DNA bases before significant DNA strand breaks appear, indicating that reactive oxygen species may be a contributory factor in the mechanism by which seizures cause cell death and bezafibrate by anti-oxidative activity prevent such DNA damage.
In many brain regions, there are expression of PPAR-α on the neurons.48,49 PPAR-α is activated by both endogenous ligands [N-acylethanolamines oleoylethanolamide (OEA) and palmitoylethanolamide (PEA)]50 and synthatic ligands such as hypolipidemic fibrates.51 In vitro and in vivo experiments have demonstrated that nicotine-evoked excitation of dopamine neurons as well as nicotine addictive properties in rats and monkeys, are suppressed by ligands to the PPARα.51–54 Evidences reveal the non - transcriptional interaction between PPARα and nAChR, via phosphorylation52,53 and this mechanism might account for the PPAR-α ligands induced blockade of neuronal and behavioral responses to nicotine.51,52,54 Study by Piligheddu M et al, demonstrated the antiepileptic effects of PPAR-α agonists in their study.16 In their study both the acute administration of PPAR-α agonist WY 14643 (WY 80 mg/kg, i.p.) or chronic administration of fenofibrate in diet (0.2%) for 14 days significantly reduced or abolished behavioral and EEG expressions of nicotine-induced seizures. Acute WY effects were reverted by the PPARα antagonist MK886 (3 mg/kg, i.p.). Another study by Citraro R et al, also documented the antiepileptic action of PEA through CB1 and PPAR-α activation in genetic model of absence epilepsy in rat.17 In this study, the authors administered PEA, anandamide (AEA), a PPAR-α antagonist (GW6471) and a synthetic CB1 receptor antagonist/inverse agonist (SR141716) to WAG/Rij rats in order to evaluate the effects on epileptic spike-wave discharges (SWDs) on EEG recordings. GW6471 antagonized PEA’s effects whereas it did not modify AEA’s effects. Furthermore, the authors have also measured PEA, AEA and 2-AG (2-arachidonoylglycerol) brain levels identifying significant differences between epileptic and control rats such as decreased PEA levels in both thalamus and cortex that might contribute to absence epilepsy. Therefore, these data demonstrate that PEA has anti-absence properties in the WAG/Rij rat model and that such properties depend on PPAR-α and indirect activation of CB1 receptors.17
The underlying mechanisms of anticonvulsive effects of Bezafibrate such as PPAR-α activation, which in turn is responsible for activation of several pathways and other mechanisms, should be investigated. Further studies are needed to explore its roles. If our data can be confirmed by clinical trials, the management of dyslipidemia may be modified, in particular for epileptic patients.
To conclude these findings may provide insights into the understanding of the mechanism of bezafibrate as an anti kindling agent and could offer a useful support to the basic antiepileptic therapy in preventing the development of PTZ induced seizures, suggesting its potential for therapeutic applications in temporal lobe epilepsy.