The Role of Focal Epilepsy Features in Defining SCN1A Mutation-positive Dravet Syndrome as Generalized and Focal Epilepsy
Article information
Abstract
Background and Purpose
This study was aimed to describe focal epilepsy features of SCN1A mutation-positive Dravet syndrome patients.
Methods
A total of 82 SCN1A mutation-positive patients were reviewed retrospectively (39 boys and 43 girls). Seizure type and electroencephalography (EEG) findings were investigated according to the stage, disease onset, and steady state (after age 2 years). Long-term video EEG data were used to classify the seizure type.
Results
Focal seizures at onset and the steady state were found in 54.9% (45/82) and 90% (63/70) of patients, respectively. Afebrile focal seizures were an initial seizure in about one fourth of the patients (22/82, 26.8%). Of 48 seizures captured during long-term video EEG monitoring of 30 patients, 19 seizures were classified as focal onset (39.6%). Of the 19 focal seizures, 12 were either focal motor or focal non-motor seizures, and seven were focal onset bilateral tonic-clonic seizure. Focal epileptiform discharges were more frequent than generalized epileptiform discharges at seizure onset and during the clinical course on conventional EEG (3.7% vs. 0%, 52.9% vs. 32.9%, respectively).
Conclusions
Our study provides a comprehensive description of focal epilepsy features of SCN1A mutation-positive Dravet syndrome patients. Recognizing these features as defining the clinical spectrum of Dravet syndrome may lead to earlier genetic diagnosis and tailored management.
Introduction
After the first description by Charlotte Dravet, Dravet syndrome has been recognized as a synonym involving severe myoclonic epilepsy of infancy.1 However, borderline phenotypes such as the absence of myoclonic seizures or absence of generalized epileptiform discharges have been consistently reported.2–4 Moreover, there are no significant differences in clinical outcome and SCN1A mutation rate between the classic and borderline phenotypes.5,6 Consequently, Dravet syndrome has replaced severe myoclonic epilepsy of infancy and now includes both the classic and atypical or borderline phenotypes.
The change in the concept underlying the classification of epilepsy syndromes as recently proposed is the introduction of epilepsy syndrome combined with both generalized and focal epilepsy.7 Dravet syndrome and Lennox-Gastaut syndrome were listed as examples of this type of generalized and focal epilepsy syndrome.7,8 In the 1989 International League Against Epilepsy (ILAE) proposal for the classification of seizure and epilepsy, severe myoclonic epilepsy in infancy was listed as “epilepsies and syndromes undetermined as to whether they are focal or generalized”.9 Recognition of unilateral clonic or alternating hemiclonic seizure at onset and later occurrence of other types of focal seizures may have prevented this epilepsy syndrome from being classified as generalized epilepsy. Earlier studies reported that focal seizures occur in 43–78.6% of Dravet syndrome patients.4,5,10 However, the presence of SCN1A mutations in these cohorts was confirmed in only some patients. Recent studies of large cohorts of SCN1A mutation-positive Dravet syndrome patients also reported the occurrence of focal seizures in 50–80.4% of patients.11–13
Nevertheless, we believe focal epilepsy features should be investigated comprehensively to be able to define Dravet syndrome more precisely as a generalized and focal epilepsy syndrome for the following reasons. First, this syndrome should be described within a homogeneous cohort of patients with SCN1A mutation. Dravet syndrome with a genetic cause other than SCN1A has been increasingly reported, and there is a concern that the clinical features and outcomes may differ according to the genetic cause.14 Second, given the evolving nature of Dravet syndrome from seizure onset to steady state, the focal epilepsy features should be described according to the clinical stage. Third, because most seizure types are determined based on the caregiver’s description, data from video electroencephalography (EEG) are needed to confirm the type of seizures.
In the present study, we investigated features of focal epilepsy in Dravet syndrome patients with a pathogenic variant of SCN1A. We discuss the relative contribution of focal epilepsy to defining the clinical spectrum of SCN1A mutation-positive Dravet syndrome.
Methods
Subjects
This study was approved by the Institutional Review Board of Seoul National University Hospital (1504-092-666). A total of 82 patients with pathogenic SCN1A variants who met the 1989 criteria for Dravet syndrome proposed by the ILAE were enrolled retrospectively in this study from four pediatric neurology centers. Briefly, all patients had seizure onset before 12 months of age, one or more generalized or focal seizures, and a variable degree of developmental delay or intellectual disability. We excluded the patients who had seizure onset after 12 months of age or normal psychomotor development within the second year of life.
Clinical data abstraction
Clinical and demographic data were collected from electronic medical records by two authors (Y.J.K and I.H.Y). We considered the stage after the age of 2 years to represent the steady state which is characterized by multiple type of seizures and slowing of development.15,16 Among 82 patients, 70 patients met this criterion. Data including age at onset, seizure type, duration of seizures, presence of fever, vaccination history, resistance to antiepileptic drugs, and developmental delay were collected. The seizure types at onset and during the steady state were classified based on the caregiver’s description. Vaccination-related febrile seizure was defined as having occurred within 48 hours of a vaccine.17 Prolonged seizure was defined as a seizure duration >15 minutes. Drug resistance was defined as failure in trials of two tolerated, appropriately chosen and used antiepileptic drug schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom.18
Interictal EEG and prolonged video EEG data
Interictal EEG recordings were obtained with 19 electrodes according to the International 10–20 system during sleep only or both during wakefulness and sleep according to the patient’s age and cooperation. The EEG data were analyzed to identify abnormalities of background activity and focal or generalized epileptiform discharges. Thirty patients underwent routine 21-channel prolonged video EEG at a sampling rate of 256 Hz with a filter setting of 1–70 Hz. All electrodes were placed according to the International 10–20 system. Data including age at the time of examination, semiology of seizure and ictal EEG pattern were collected. The semiology of seizure and ictal EEG pattern were analyzed by professional epileptologists at each center.
Genetic analysis
The SCN1A pathogenic variants were identified from various genetic tests: Sanger sequencing (56 patients), gene panel (22 patients), and multiple ligation-dependent probe amplification (4 patients). We classified the SCN1A variants as pathogenic if reported in either ClinVar or Human Gene Mutation Database as pathogenic or disease-causing mutations. For novel missense variants not reported in a population database (gnomAD or ExAC), we considered the variant as pathogenic only if the variant was confirmed as de novo or segregated in the affected family member. For the novel loss-of-function variant (nonsense, frameshift indels, splice site), we considered the variant as pathogenic regardless of the family test result. For the Ko YJ, et al. Focal Epilepsy Features in SCN1A Mutation-positive Dravet Syndrome deletions involving multiple exons of SCN1A, we included only those variants that do not span the adjacent sodium channel genes SCN2A and SCN3A. Our strategy met the criteria for defining a mutation as pathogenic or likely pathogenic suggested by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.19
Results
The study cohort included 39 boys and 43 girls. The 80 SCN1A pathogenic variants are summarized in the Table 1. In brief, 13 nonsense, 20 small insertion/deletions, six splice site variants, 37 missense variants, and four large deletions were identified. Twenty pathogenic sequence variants have not been reported in the literature or variant databases (ClinVar or Human Gene Mutation Database).

The 80 SCN1A variants found in 82 patients
At onset
The mean age at seizure onset was 5.3 months (range, 1 day–11.5 months). The seizures at onset were prolonged (>15 minutes) in 24 patients (24/82, 29.3%). Four patients experienced their first seizure within 48 hours after vaccination, and fever was associated with the seizures in two of them. In 39 patients, the initial seizures were classified as focal clonic, including hemiclonic or alternating hemiclonic (47.6%). Nonmotor onset focal seizures with behavioral arrest and autonomic symptoms were found in six patients (6/82, 7.3%). Bilateral tonic-clonic seizures suspected as having a generalized onset from the caregiver’s description were found in 35 patients (35/82, 42.7%). In two patients, generalized myoclonic seizures were suspected as the initial seizure. Fever was associated with the initial seizure in 42 patients (42/82, 51.2%). The frequencies of generalized and focal seizures at onset were similar regardless of the presence or absence of fever (Fig. 1). Interictal EEG performed at seizure onset was normal in 66 patients (66/69, 95.7%). Focal epileptiform discharges were found in three patients (3.7%), and generalized epileptiform discharges were not found in any patients.
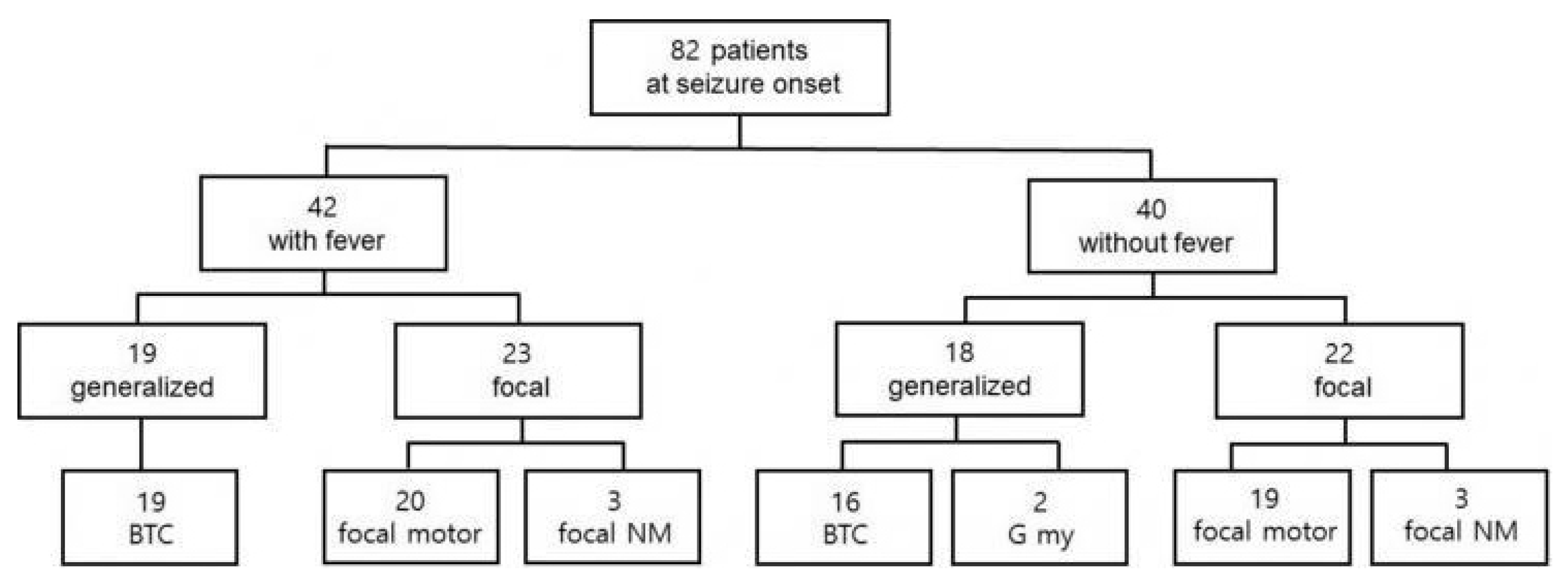
The frequencies of focal and generalized seizure at onset in the two groups classified according to the presence of fever. BTC, bilateral tonic-clonic seizure; focal NM, focal non-motor seizure; G My, generalized myoclonic seizure.
Steady state
The electroclinical data of 70 patients (31 boys and 39 girls) were available after age 2 years. The mean follow-up duration of these 70 patients was 111.4 months (range, 2–327.2 months). Fourteen of the 70 patients were followed up past the age of 18 years (average follow-up duration 265.25 months, range 220.6–327.2 months). Both generalized and focal seizures during steady state were reported in 59 patients (59/70, 84.3%). Bilateral tonic-clonic seizures were reported in 60 patients. Of these 60 patients, semiologic features suggestive of focal onset to bilateral tonic-clonic seizure were found in 12 patients. The type and frequency of focal and generalized seizures are shown in Fig. 2A. During the follow-up, four patients showed only focal seizures and seven patients showed only generalized seizures. The mean number of EEG recordings obtained for each patient during steady state was 5.0 (range, 1–20). Focal epileptiform discharges were identified in 37 patients (37/70, 52.9%), and generalized epileptiform discharges were found in 23 patients (23/70, 32.9%). Twenty-four patients showed neither focal nor generalized epileptiform discharges (24/70, 34.3%).

The frequency of seizure type during steady state. The frequency of focal and generalized seizure type was classified from the clinical description (comprising a total of 163 seizures in 70 patients) (A). The frequency of focal and generalized seizure confirmed during video electroencephalography recording (comprising a total of 48 seizures in 30 patients) (B). At, atonic; NM, FO, nonmotor, focal onset; AA, atypical absence; M, FO, motor, focal onset; BTC, GO, bilateral tonic-clonic seizure, generalized onset; BTC, FO, bilateral tonic-clonic seizure, focal onset; ES, GO; epileptic spasms, generalized onset; My, myoclonic.
Long-term video EEG monitoring
Thirty-three long-term video EEG recordings were made for 30 patients. The mean age at the time of video EEG monitoring was 4.3 years (range, 1.1–12.1 years). The types and frequencies of focal and generalized seizures captured during the video EEG monitoring are shown in Fig. 2B. Among the total of 48 seizures, 19 seizures were focal onset and 29 seizures were generalized onset. Of the 19 focal onset seizures, five focal motor seizures and seven focal non-motor seizures were captured. Seven bilateral tonic-clonic seizures classified as focal based on either an evolving ictal EEG pattern of focal to bilateral (Fig. 3A) or prominent hemispheric asymmetry during the recording (Fig. 3B). Ten bilateral tonic-clonic seizures were classified as generalized onset (Fig. 3C). Presurgical evaluation was performed in three patients and included long-term video EEG monitoring. In one patient, brain magnetic resonance imaging, positron emission tomography with 2-deoxy-2-[fluorine-18]fluoro-d-glucose, and ictal EEG findings suggested the right posterior temporal lobe as an epileptogenic zone. Resective surgery is planned after invasive monitoring for this patient.

The ictal EEGs of bilateral tonic-clonic seizures. EEG for a 12-year-old boy who showed left arm clonic movement followed by bilateral tonic-clonic movement. The ictal EEG began with irregular delta and rhythmic spike waves over the right hemisphere and ended with diffuse spike waves (A). EEG for a 6-year-old boy who showed bilateral tonic-clonic seizure, which was more prominent in the left arm and leg. The ictal EEG began with generalized spike waves. The repetitive spikes and subsequent spike waves were more prominent in the right hemisphere throughout the recording (B). EEG for a 4-year-old boy who showed bilateral tonic-clonic seizures preceded by myoclonic movements in both arms. The ictal EEG began with generalized spike waves. The rhythmic spikes and subsequent spike waves were symmetric throughout the recording (C). Longitudinal bipolar montage, sensitivity 25 μv (A), 25 μv (B), and 75 μv (C). EEG, electroencephalography.
Discussion
Our study describes the features of focal epilepsy in a large cohort of SCN1A mutation-positive Dravet syndrome patients. The frequency of focal seizures was similar to that of generalized seizures when analyzed by both clinical history and long-term video EEG monitoring. About 40% of the bilateral convulsive seizures captured during the video EEG recording revealed features of focal epilepsy. Interictal focal epileptiform discharges were detected more frequently than generalized epileptiform discharges at both seizure onset and during the clinical course. Of particular note in our study is that fever was not associated with seizure onset in up to 50% of the patients. The reported frequency of fever at onset ranges from 28% to 70% in different studies.10,20,21 In our study, the frequency of focal and generalized seizures at onset did not differ significantly according to the presence or absence of fever. Therefore, afebrile focal motor and non-motor seizures may be the initial seizure in a substantial proportion of SCN1A mutation-positive Dravet syndrome patients. The findings of these predominant features of focal epilepsy raise questions about the border zone or extent of clinical spectrum of SCN1A mutation-positive Dravet syndrome.
Previously, cryptogenic focal epilepsy6,22 and epilepsy of infancy with migrating focal seizures23 were recognized as an example of the pure focal epilepsy spectra of SCN1A-related epilepsy. Likewise, cryptogenic generalized epilepsy,6 intractable childhood epilepsy with generalized tonic-clonic seizures,24 and epilepsy with myoclonic-atonic seizures,25 which is considered to be pure generalized epilepsy, are thought to represent the opposite end of SCN1A-related epilepsy. Despite the variable follow-up duration, our study cohort showed similar tendencies; that is, the majority of patients exhibited both focal and generalized epilepsy features during their clinical course (59/70, 84.3%). However, 11 of 70 patients (15.7%) showed either pure focal (four patients) or generalized (seven patients) epilepsy. Thus, it appears that the exact boundaries defining epilepsy syndrome of SCN1A mutation-positive patients is becoming blurred in terms of the seizure type.
Even before the advent of the genomic era initiated by the development of next-generation sequencing, focal features observed in patients with infantile onset epilepsy were of interest because these focal features made it difficult to distinguish cryptogenic localization-related epilepsy from severe myoclonic epilepsy in infancy.26 However, if SCN1A genetic testing had been performed in this cohort, SCN1A mutation may have been identified in both epilepsy syndromes. Other sodium channelopathies including SCN2A and SCN8A have also been reported to present with focal seizure during infancy.27–29 Interestingly, some SCN2A- or SCN8A-related epilepsy patients have shown a favorable response to sodium channel blockers, which are not recommended for SCN1A-positive Dravet syndrome patients because of possible seizure aggravation.30 The pathophysiological mechanism explaining different responses according to the mutated sodium channel subunits has not been elucidated. However, prompt genetic diagnosis at onset may guide the use of sodium channel blockers according to the mutated sodium channel subunits.
Our study has several limitations because it was a retrospective study. First, seizure type classification was made based on a retrospective review of medical records. The differentiation of focal and generalized features could have been weighted differently by the attending clinician, and the relative frequencies of focal and generalized seizures in our study should be compared with another large retrospective cohort or a prospective cohort. Second, we defined the broad range of steady state (after age 2 years) because of the variable duration of the follow-up in these patients. Longer follow-up and further differentiation of the steady state into early childhood, adolescence, and adulthood may provide additional information about the evolution of seizure type. Nevertheless, we believe that our video EEG monitoring results for 30 patients partially compensate for these limitations by providing objective data about the seizure type during childhood.
In summary, our study describes the focal features of 82 SCN1A mutation-positive Dravet syndrome patients. Afebrile focal seizures were the initial seizure in about one-fourth of the patients. The frequency of focal seizures was similar to that of generalized seizures at seizure onset and during the steady state. Focal epileptiform disKo charges were found more frequently than generalized epileptiform discharges both at seizure onset and during the steady state. Video EEG monitoring data showed that focal seizure onset accounted for 40% of the recorded seizures. These comprehensive data will provide solid evidence for defining SCN1A mutation-positive Dravet syndrome as a type of generalized and focal epilepsy.
Notes
Conflict of Interest
The authors declare that they have no conflicts of interest.