Isolated Unilateral EEG Findings in Juvenile Myoclonic Epilepsy: A Case Report
Article information
Abstract
Juvenile myoclonic epilepsy (JME) has well-defined clinical and electrophysiological features. On the other hand, large case series have shown that focal and asymmetrical discharges may accompany generalized epileptiform activities in JME. Although it is known that these non-generalized electrophysiological findings do not exclude the diagnosis of this syndrome, some findings may create confusion in the differential diagnosis. In this case report, a case of JME with electroencephalographic findings characterized by isolated unilateral epileptiform activities without typical generalized discharges was discussed. The current case clinically presented with involuntary jerk movements in the bilateral upper extremities. It has been determined that these movements are uni/bilateral myoclonic beats based on home video recordings. Metabolic, toxic and structural problems were excluded in the investigations for the etiology of myoclonus. In the electrophysiological examination performed for epileptic processes, epileptiform discharges localized to the isolated right hemisphere were observed. JME was considered primarily due to clinical findings in the patient, and effective seizure control was achieved in a 4-year follow-up under anti-seizure treatment. The peculiarity of the case is the presence of electrophysiology recordings of isolated unilateral epileptiform activity during the 4-year follow-up period. It should be emphasized that there is no case of JME diagnosed with isolated unilateral epileptiform activity in the absence of generalized spike-slow waves or multiple spike-slow waves in the literature.
Introduction
Juvenile myoclonic epilepsy (JME) is an idiopathic generalized epilepsy that is common in adolescent and young adult age, with a prevalence of 5–10% among all epilepsy cases.1 The clinical and electro-encephalographical (EEG) features were first described by Janz and Christian in 1957.2 JME typically presents with uni- or bilateral arrhythmic, irregular single myoclonic beat or clustered beats3 with preserved consciousness.4 In addition to myoclonic beats, 85–100% of cases have generalized tonic-clonic seizures, 20–40% have absence seizures, and rarely convulsive, non-convulsive or absence status epilepticus.5,6
The well-defined EEG features of JME can be listed as: ground activity is expected to be normal.7 During interictal period, generalized spike-slow wave (SSW) or multiple spike-slow wave (MSSW) activity of >3.5 Hz, triggered by intermittent photic stimuli, hyperventilation and sleep-deprivation with increased frequency during and after sleep, is observed.7 In the ictal period, after 2–5 seconds of generalized SSW or MSSW activity, myoclonic discharges appear within 20–50 miliseconds.7 In addition to these electrophysiological features, various focal and asymmetric findings can be detected in JME.8,9 In this study, a JME case with persistent isolated unilateral EEG findings is discussed which has not been described in the literature yet.
Case Report
The patient, who was completely healthy regarding neurocognitive development and without any history of seizures or febrile convulsions before, applied to our department at the age of 20. He had involuntary movements in his right arm, which had been almost every day for about 1 year. This movement was described by the patient as the right shoulder being thrown forward from the trunk and the arm suddenly thrown forward, “like a boxer's punching motion”. These movements became more frequent in the early post-sleep period and caused the patient to drop the objects in his hand and difficulties in fine motor skills. The involuntary beats were not accompanied by loss of consciousness. The patient did not have a suspicious history in terms of cognitive decline or attention deficit. There was no significant pathology in the medical history of the patient and his family. Neurological examination was normal and his cognitive status was appropriate for his age. When the home video of the attacks was examined, myoclonic beats in both upper extremities simultaneously, sometimes affecting both lower extremities, causing the patient to drop the phone he was holding in his hand and stagger while standing, were noted in the first half hour after awakening.
Any pathological findings were not detected in cranial magnetic resonance imaging performed for possible structural problems that may lead to myoclonia (Fig. 1). Toxic and metabolic disorders that can be associated with myoclonus were excluded. Although there was no history of generalized seizures, JME was placed at the top of the differential diagnosis list and the patient underwent EEG. While the ground activity was normal 8–13 Hz alpha rhythm in the left hemisphere, it was significantly slowed in the right hemisphere. Intermittent photic stimulation was within normal limits. While left hemisphere activity was normal in hyperventilation, diffuse SSW was present in the right hemisphere. The frequency of right hemispheric SSW increased significantly during sleep and this activity was limited only to the right hemisphere (Fig. 2). During the EEG, any myoclonic or ictal activities could not be recorded.
Non-contrast cranial MRI examination within normal limits. Sections (A) axial T2/FLAIR-weighted, (B) axial T2-weighted, (C) coronal T2-weighted, and (D) sagittal T1-weighted. MRI, magnetic resonance imaging; FLAIR, fluid attenuated inversion recovery.
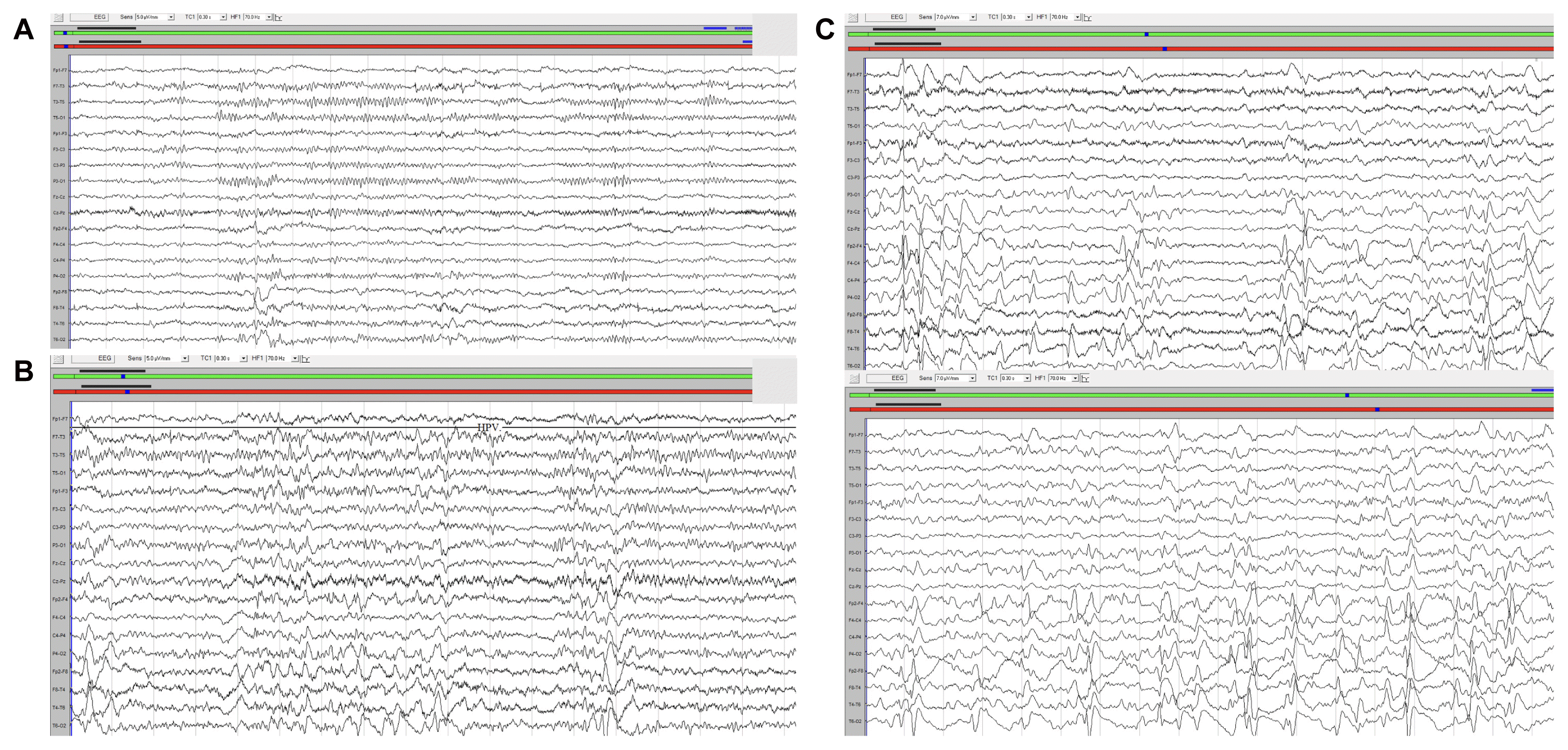
Isolated unilateral EEG findings of the patient. (A) Asymmetric slowing of the ground activity in the right hemisphere in wakefulness EEG. (B) Right hemisphere spike-slow wave paroxysms during hyperventilation. (C) Right hemisphere spike-slow wave paroxysms during sleep. EEG, electroencephalography.
Considering the absence of any structural pathology that could explain the unilateral EEG findings in the patient's cranial imaging, the clinical and EEG features were associated with a generalized epilepsy syndrome. With the preliminary diagnosis of JME, the patient was started on lamotrigine (initial dose was 12.5 mg/day, with gradual weekly dose increases of 12.5 mg, maintenance dose was reached to 100 mg/day). The patient, whose myoclonic pulses ended under this treatment, developed a generalized tonic-clonic epileptic seizure for the first time during a period when anti-seizure medication was interrupted. Repeated EEGs showed similar findings with the first one. Lamotrigine was again increased to 100 mg and the patient was followed for 4 years under this treatment. At the end of this period, repeated EEG findings were similar to the first ones. The patient did not develop any generalized seizures during the 4-year period. It was planned to continue the current treatment of the patient since myoclonia developed in his upper extremities during periods of drug discontinuation when he was sleep deprived or played video games for a long time.
Discussion
Although generalized SSW or MSSW that can be triggered by photic stimuli and increase in frequency with hyperventilation or sleep are well-defined electrophysiological anomalies, different focal and asymmetrical EEG findings may also occur in JME. The frequency and different types of focal and asymmetric EEG findings in JME were first documented in 1994.10 In this study, 49 EEG of 22 JME cases were examined and focal abnormalities were detected in 18 EEG of 12 patients. In 13/18 EEGs with focal abnormalities, slow wave foci that develop independently from generalized discharges were shown and in five of them the focus persisted in one hemisphere. After this first study, focal and asymmetrical features observed in the EEG of JME cases were discussed in many different publications.8,11,12 In these publications, generalized SSW and MSSW accompanying amplitude asymmetry and focal onset generalized discharges were seen in JME cases between 10% and 44.6%.8,11,12 The focal anomalies occurring independently of generalized discharges ranged from 6.6% to 32.6%.8,11 Independent focal EEG abnormalities were listed as frontal, temporal, occipital, and parietal sharp/spiky waves, phase reversals, or intermittent rhythmic delta activities in the frontal and occipital regions.8
In a study examining the resistance to anti-seizure drugs, no relationship was found between the presence of asymmetric or focal features in EEG and resistance to treatment, and EEG features did not have an effect on the response to anti-seizure drugs.13 However, it should not be overlooked that all cases examined in this study were treated with broad-spectrum anti-seizure drugs (valproate, lamotrigine, topiramate and levetiracetam) despite their asymmetric or focal EEG features. It is known that asymmetric or focal EEG findings lead to misdiagnosis, and 30–40% of JME cases is misdiagnosed as focal epilepsy.14 In the literature, there are JME cases with an increased frequency of seizures and even diagnosed with treatment-resistant focal epilepsy while being followed up under carbamazepine due to asymmetric or focal EEG findings.15
The presented case of JME is unique in that isolated unilateral epileptiform discharges were detected rather than generalized SSW or MSSW discharges. This case suggests the diagnosis of JME clinically due to his seizure semiology and the exclusion of secondary etiologies. However, electrophysiological suspicion persists because of the unilateral and focal findings localized only to the right hemisphere and the failure to show generalized SSW or MSSW. Although it is not possible to make a definitive diagnosis of JME considering the current criteria, the case can be evaluated in favor of JME due to clinical presentation and effective anti-seizure treatment response. Therefore, the presence of isolated asymmetric or focal EEG abnormalities should not exclude the JME, and patients should be evaluated by both the clinical findings and electrophysiological features during untreated period and under appropriate anti-seizure treatment. Diagnosis of JME should not be delayed due to unusual EEG findings and appropriate anti-seizure medications should be started immediately.
Notes
The authors declare that they have no conflicts of interest.