The administration of vigabatrin (VGB), an irreversible gamma-aminobutyric acid (GABA)-transaminase inhibitor, into thalamocortical relay neurons (TCRs) may increase spontaneous cortical spike-and-wave discharges in the rat.1 Using VGB also may increase the frequency and severity of absences and absence status in the patients with idiopathic generalized epilepsy.2,3 In addition, VGB may induce de novo absence status or de novo absence seizures with generalized epileptiform discharges in the patients with focal epilepsy.4,5
This report is about a patient with focal epilepsy whose seizure patterns are occipital lobe onset clinically. In the patient, VGB induced generalized epileptiform discharges were observed in consecutive EEGs taken over approximately four years. However, there was no definitive provocation of primary generalized seizures.
Case Report
A 21-year-old man has been treated for a seizure disorder in the Department of Neurology since he was 15 years old. At the age of nine years old, he suffered the first seizure attack. The beginning of his seizures was a visual aura, right side hemianopsia, usually accompanied by a headache, and then he stares vacantly and does not respond to verbal commands. The frequency of the seizures was two to three times per year.
When he was 13 years old, he had started to take an unknown species of antiepileptic drug. But because of the bankruptcy of the hospital he was treated in, he stopped the medication when he was 14 years old. He suffered a generalized tonic-clonic seizure under the drug withdrawal state. Just after the seizure he visited another hospital. Valproic acid (VPA) monotherapy did not change the seizure frequency under poor drug compliance. He heard that there were no abnormal findings on the two EEGs checked in that hospital.
The patient visited the pediatric department of our hospital when he was 15 years old. There were no abnormal findings in the brain MRI taken at that age; VGB was added on and VPA was tapered; his prescription was adjusted to high dosage of VGB (3,500 mg/day) by a pediatrician. Five months after his visit to the pediatric department, he suffered another complex partial seizure, and the pediatrician in charge referred the patient to the department of Neurology. Since then, he had been seizure free for four years and eight months while taking 3,000 mg/day of VGB. He also did not report any absence attack during that period. However, he had another complex partial seizure starting with visual aura when he was 20 years old; that was his last seizure.
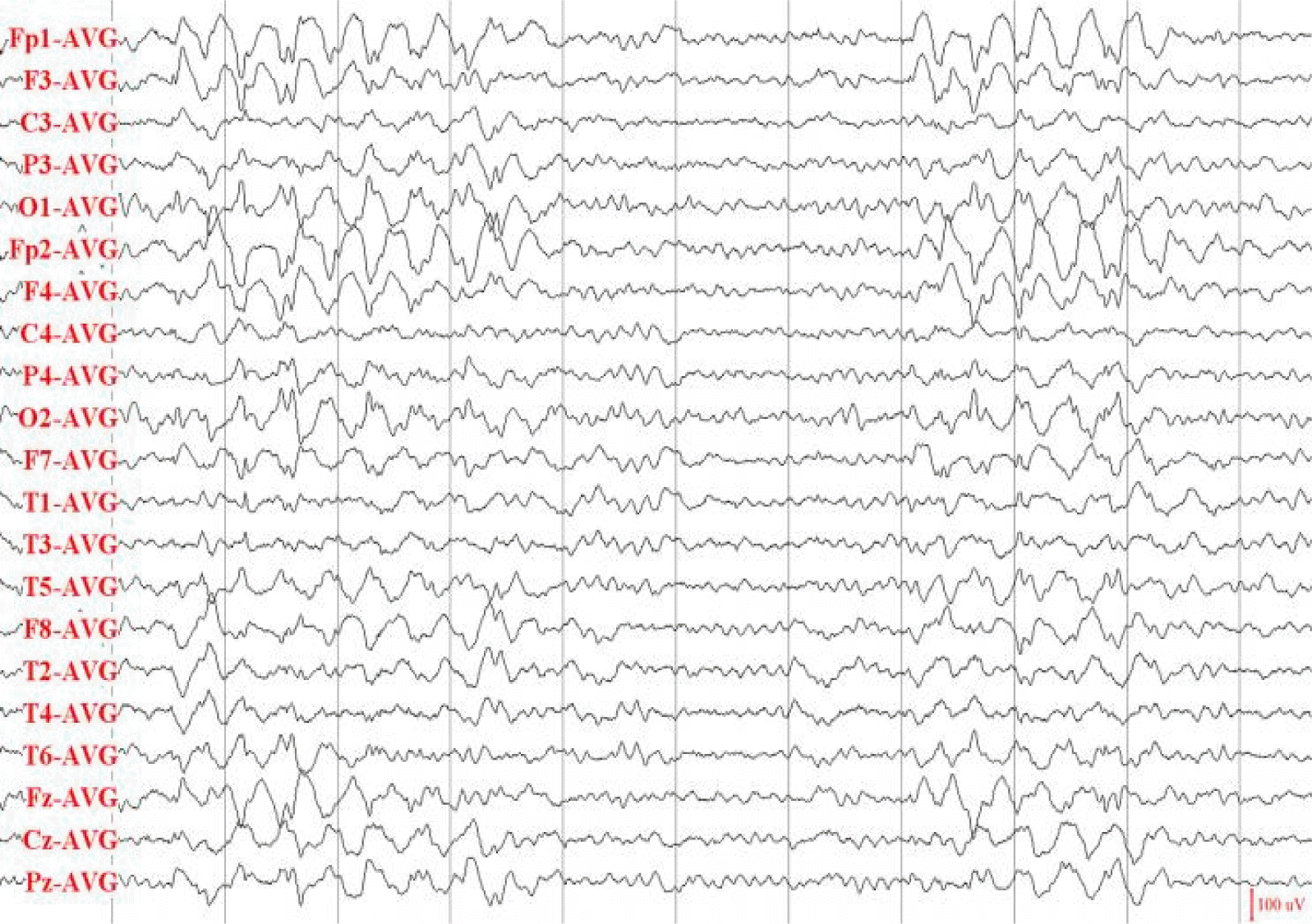
EEGs were recorded periodically in our hospital after the use of VGB since the first visit in the pediatric department. In eight of the consecutive nine EEGs recorded over approximately four years, the diffuse bursts of 2–2.5 Hz spike-and-wave complexes were observed repetitively (Fig. 1). The occurrences of the abnormal discharges in each EEG were several to frequent during the 30 min recording. In another EEG recorded five months after the last seizure at when he was 20 years old, the generalized epileptiform discharges were also observed intermittently. Just after that EEG recording, 600 mg/day of VPA was added to his medication, and the tapering of VGB was started.
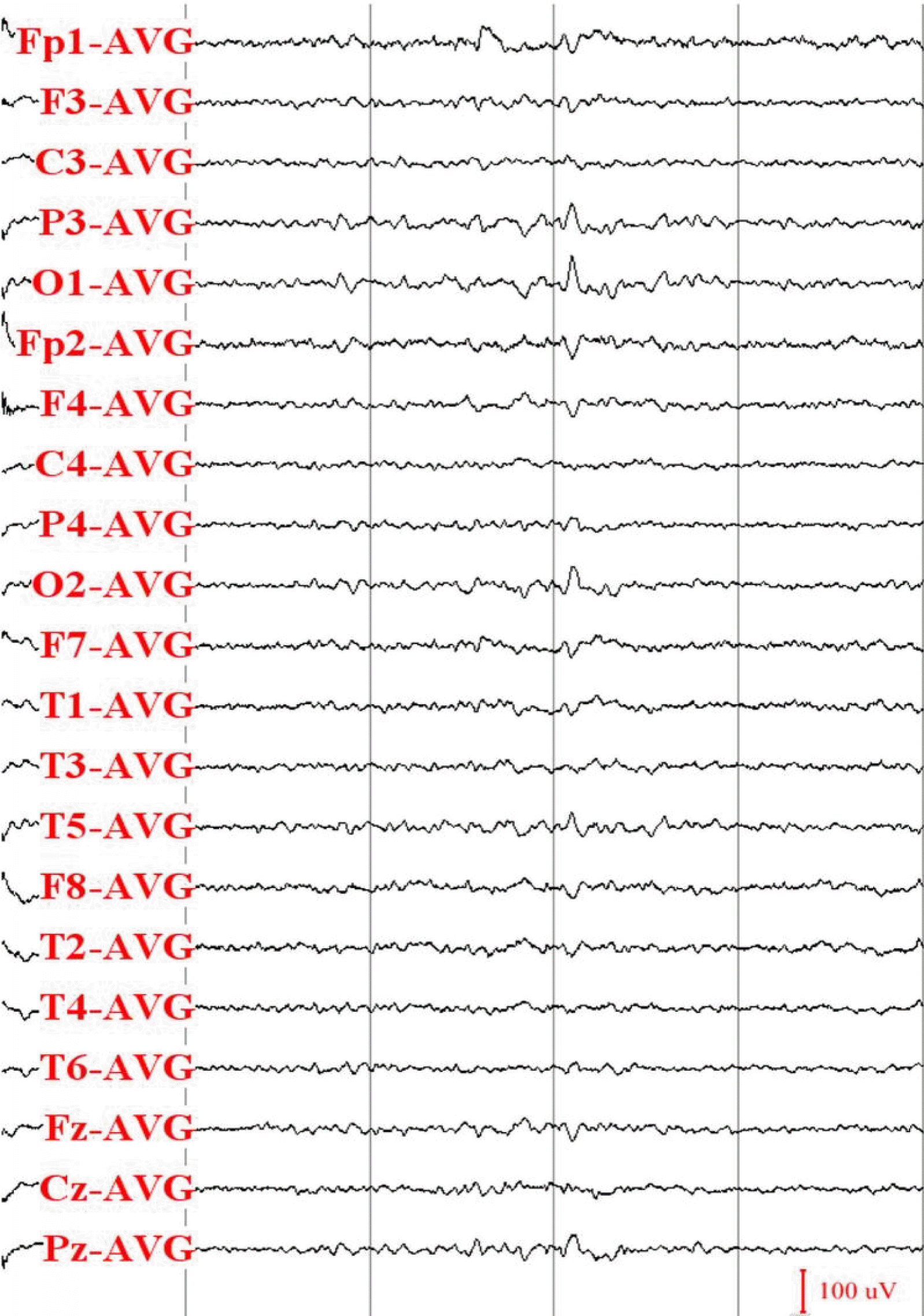
Four months after starting of the drug changes, another EEG was done. The patient’s prescription at that time was 600 mg/day of VPA plus 1,000 mg/day of VGB. In the EEG recorded under that medication, several clusters of spikes on the occipital area of the left hemisphere were observed (Fig. 2). There was no burst of the 2–2.5 Hz spike-and-wave complexes on that EEG. One month after the EEG recording, the VGB medication was stopped after tapering. When the patient was 21 years old, four months after the EEG recording, another EEG was recorded; no epileptiform discharges were observed in that EEG. The medication at that time was 600 mg/day of VPA.
Discussion
The mechanism of VGB induced-seizure aggravation is thought to associate with GABAergic system that VGB may act antiepileptic role. In the thalamocortical circuit, nucleus reticularis thalami neurons (NRTs) are GABAergic neurons and the pacemakers generating spindle waves. The inhibitory impulses produced by the firing of NRTs keep TCRs in an inhibitory state. The overactivation of the GABAB receptors of the TCRs, resulting from the exceedingly active state of NRTs, changes the spindle waves to the spike-and-wave-complexes.6
Enhancement of GABAergic transmission could show both preventive and aggravating effects to seizures depending on the site of action. The systemic or local GABAergic stimulation by drugs increased the occurrences of seizures and the duration of spike-and wave discharges in pharmacological and genetic animal models of absence seizures.7,8 In the microinjection study with the genetic absence epilepsy rats from Strasbourg (GAERS), the injection of VGB into TCRs increased spontaneous cortical spike-and-wave discharges but the injection into NRTs inhibits the discharges. This may be because the inhibition of NRTs reduces the tendency of synchronization of TCRs, but the enhanced inhibition of TCRs by VGB may make the TCRs in a more synchronized state.1,9
Worsening of seizures induced by VGB has been reported usually in the patients with generalized epilepsy.2,3 VGB also could induce de novo absence seizures or absence status epilepticus with generalized epileptiform discharges in patients with partial seizures.4,5 In addition, myoclonic jerks with generalized polyspike and wave complexes in EEG were observed during VGB therapy in focal epilepsy.10 However all of seizures induced by VGB, in both of the aggravated cases and the de novo cases, subsided after the discontinuation of VGB or switching from VGB to other antiepileptic drugs.4,5,10
The VGB-induced generalized spike-and-wave complexes had been observed for about four years in this patient, who had focal epilepsy; there was no clinically definitive provocation of primary generalized seizures. It is certainly difficult to monitor the occurrence of absence seizures completely. However he had never reported about absence seizures despite repetitive inquiries. After the introduction of VPA and reduction of VGB, there was disappearance of the generalized epileptiform discharges completely. The pattern of epileptiform discharges on that EEG was focal type, which confined to the occipital area of the left hemisphere. There was no epileptiform discharge in another EEG taken after the VGB withdrawal.
The use of VGB may chronically induce generalized epileptiform discharges without the induction of primary generalized seizures in the patients with focal epilepsy. Regardless that it is not clear whether the discontinuation of VGB, the introduction of VPA or a combination of both disappear the generalized epileptiform discharges in this case. The discontinuation of VGB would be helpful to suppress the generalized epileptiform discharges in such cases.