Chromosome 22q11.2 deletion syndrome is the most common interstitial deletion syndrome, with an incidence of 1 in 4000–5000 births and approximately 5–10% with inherited deletions.1 Initially, this condition was referred to as CATCH22, the acronym of cardiac defect, abnormal face, thymic hypoplasia, cleft palate, and hypocalcemia.2 However, information from recent genetic studies proves that the chromosome 22q11.2 deletion syndrome includes DiGeorge syndrome, velo-cardio-facial syndrome, and conotruncal anomaly face syndrome.1 At least 10% of patients with this syndrome were reported to have hypocalcemic seizures. Hypocalcemic seizures are rare clinical manifestations after the neonatal period because of the increase of parathyroid gland hypertrophy and dietary calcium intake.3
We hereby present two cases of the chromosome 22q11.2 deletion syndrome diagnosed in 12-year-old boys with new onset hypocalcemic seizures, one of who had a paternal deletion of the same chromosomal site. This case report hopes to call attention to this syndrome as a potential cause of hypocalcemic seizures even after the neonatal period. And this is the first case report of hypocalcemic seizure with a paternally inherited 22q11.2 deletion in Korea.
Case Report
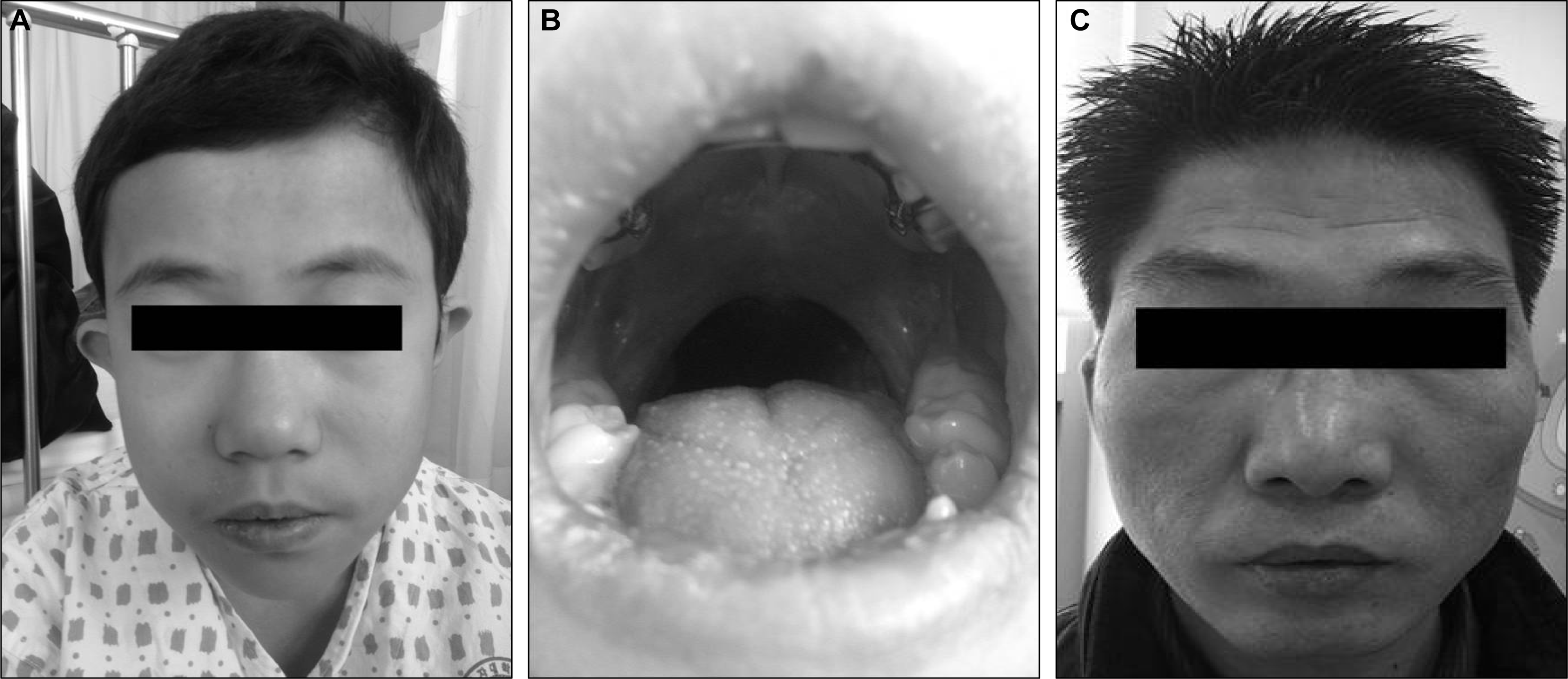
Case 1
A 12-year-old boy presented with a new onset generalized tonic-clonic seizure. He is the first child of a phenotypically normal mother. The patient was born at term after an uneventful pregnancy by an uncomplicated caesarean section with low birth weight of 2,900g (10–25th percentile). At 8 months the child underwent surgical repair of a cleft palate and at age 2 years had two episodes of febrile convulsion. He had a learning disability in his school-aged years. On family history, his father had a prominent forehead, small down-turned mouth, micrognathia, and nasal voice (Figure 1C). He had not undergone any prior specific evaluation, but seemed to have normal intelligence without any neuropsychiatric symptoms including a seizure history.
Physical examination revealed prominent forehead, small down-turned mouth, micrognathia (Figure 1A), and repaired cleft palate with not well formed uvula (Figure 1B). His height was 146 cm (25–50th percentile) and weight was 38.5 kg (25–50th percentile). Chvostek’s and Trousseau’s signs were negative. He had mild mental retardation with an intelligence quotient (IQ) of 68 on the Korean version of Wechsler Intelligence Scale for Children (K-WISC).
The electrocardiogram (ECG) was abnormal due to a prolonged corrected QT interval and the chest radiograph failed to show a thymus shadowing. Computed tomography (CT) of the heart revealed right aortic arch (Figure 2), and CT of the brain showed multiple calcifications in the bilateral basal ganglia and both frontal white matters (Figure 3A). The electroencephalogram (EEG) revealed generalized intermittent slow waves without any epileptiform discharges.
Laboratory tests showed hypocalcemia (calcium 6.5 mg/dL, ionized calcium 2.8 mg/dL), hyperphosphatemia (9.1 mg/dL) and normal level of magnesium (1.8 mg/dL). Parathyroid hormone (PTH) was decreased to 4.76 pg/mL (reference range 15–65 pg/mL) and 25-vitamin D3 was decreased to 13.6 ng/mL (reference range 30–60 ng/mL). Peripheral blood count, hepatic, renal and thyroid function tests were all normal except thrombocytopenia (144,000/μL). Immunological studies showed normal levels of Immunoglobulins but T cell subpopulation analysis was abnormal. T lymphocyte was decreased to 42.8 %/μL (reference range 52–78%/μL), CD4/CD8 T cell ratio was decreased to 0.79 (reference range 0.8–3.0).
Fluorescent in situ hybridization (FISH) analysis was performed using peripheral blood and chromosomal study was carried out using DiGeorge/VCFS TUPLE1 region probe (22q11.2) with an ARSA control probe (22q13) which confirmed a 22q11.2 deletion. His father revealed the same chromosomal abnormality, which suggests paternal inheritance in this case.
The patient was treated with calcium citrate and calcitriol. He remained asymptomatic with normalization of his serum calcium level with medications.
Case 2
A 12-year-old boy presented with a new onset generalized tonic-clonic seizure. He is the second child of phenotypically normal parents, born at term after an uneventful pregnancy by an uncomplicated caesarean section with low birth weight of 2,800 g (10–25th percentile). At age 1 year, the child underwent surgical repair of patent ductus arteriosus. At age 6 and 7 years, he had two episodes of febrile convulsion. He had a learning difficulty in his school-aged years. He did not have any family history of similar clinical conditions.
Physical examination was abnormal due to a prominent forehead, small down-turned mouth, micrognathia and bifida uvula. His height was 155.8 cm (90–97th percentile) and weight was 59.7 kg (75–90th percentile). Chvostek’s and Trousseau’s signs were negative. On neurological evaluation, his verbal fluency was decreased.
The ECG showed a prolonged corrected QT interval and chest X-ray failed to show a thymus shadowing. The brain CT revealed calcification located in the left basal ganglia (Figure 3B). The EEG was abnormal due to generalized intermittent slow waves without any epileptiform discharges.
Laboratory tests showed hypocalcemia (calcium 6.9 mg/dL, ionized calcium 3.8 mg/dL), hyperphosphatemia (8.3 mg/dL) and normal level of magnesium (1.7 mg/dL). PTH was normal with 21.91 pg/mL (reference range 15–65 pg/mL) and 25-vitamin D3 was decreased to 22.9 ng/mL (reference range 30–60 ng/mL). Peripheral blood count, hepatic and renal function tests were all within normal range except mild thrombocytopenia (130,000/μL). A 22q11.2 deletion was confirmed by a FISH analysis.
The patient was treated with calcium citrate and calcitriol. He remained asymptomatic with normal serum calcium level with medications.
Discussion
The first patient had cleft palate at birth and learning difficulty at school-aged years. At age 12 years generalized tonic clonic seizure developed. Also, hypocalcemia, characteristic face, T cell defect were observed by physicians. It turned out his father had the same chromosomal abnormality, which suggests that it was paternally inherited in this patient. The second patient had similar clinical features except the patent ductus arteriosus at birth instead of the cleft palate without any family history. Clinical absence of the typical hallmarks of this syndrome should not exclude this disease due to variable phenotypes. Continuous efforts to define diverse clinical manifestations of this syndrome will lead to proper evaluation and intervention for patients. Fortunately, the treatment of hypocalcemic seizure is relatively simple and can provide definite clinical improvement.4
A cohort study showed that 21% of the patients with chromosome 22q11.2 deletion syndrome had seizures, and at least 68% of them had hypocalcemic seizures. Most of the patients are diagnosed during the neonatal period, when the transport of calcium from mother to fetus is abruptly interrupted. The calcium level usually improves over the first year of life because of parathyroid gland hypertrophy and dietary calcium intake. For this reason, older patients seldom require ongoing calcium supplementation.4 Generally, we consider hypocalcemia as a result of the this syndrome when the patient presents symptoms such as seizure during the neonatal period especially after 72 hours of life,9,10 which is summarized in Figure 4. However, there was a case report of this syndrome in Korea before, in which the patient had hypocalcemic seizures at the age of 13 years without any family history.5 Our case report also supports that manifestation of hypocalcemic seizures beyond the neonatal period should be considered as a possible symptom of this syndrome.
Haploinsufficiency loci 22q and 10p could be associated with similar clinical features characterized by absent or hypoplastic thymus and parathyroid, and conotruncal heart defects and urinary tract abnormalities. Heterogeneity of the heart defects and abnormalities of the urinary tract are more common in patients with partial monosomy 10p than in patients with partial monosomy 22q.6 Our patients were more compatible with partial monosomy 22q because there were no evidence of urinary tract involvement, although one of them had a structural heart defect (patent ductus arteriosus). The 22q11.2 deletions were confirmed by FISH analysis in both cases.
Even though published estimates vary, about 5–10% of patients have been reported to have inherited deletions.1 A collaborative study showed 72% (204/285) of patients had de novo deletions since their parents did not have deletions on both sides. Inherited maternal deletions (77%, 61/79) were predominant compared with paternal deletions (23%, 18/79).3 Our first patient showed inheritance from a paternal deletion which is not as common as maternal deletions. We noticed that the father of this patient had similar face morphology as described earlier but he had never developed hypocalcemic seizures. He was eventually diagnosed with this syndrome by FISH analysis after his son was confirmed of this diagnosis. This is the first case of hypocalcemic seizure confirmed as the paternally inherited 22q11.2 deletion in Korea.
Intracranial calcification with the chromosome 22q11.2 deletion syndrome is not common in individuals with symptomatic hypocalcemia. Possible explanations include low incidence of this finding, or the young age at diagnosis which precludes the development of intracranial calcification, or the fact that not all patients receive brain CT.7 Our patients showed calcification in bilateral or unilateral basal ganglia and both frontal white matters on brain CT accompanied by hypocalcemic seizures at 12 years of age. An association between intracranial calcification and neuropsychiatric symptoms has been reported in familial idiopathic basal ganglia calcification.8 Further investigations would be needed to determine the significance of the intracranial calcification with direct associations with neuropsychiatric symptoms in this syndrome.