Introduction
Dyke-Davidoff-Masson syndrome (DDMS) was originally characterized by its radiologic features, which include cerebral hemiatrophy and ipsilateral skull hypertrophy with hyperpneumatization of the paranasal sinuses and mastoid cells.1,2 Although seizure is one of the core symptoms along with hemiparesis and mental retardation, few papers described long-term course. We present one typical and one suspicious case within one family and summarize published cases concerning seizures. In addition, the plausible causes of familial occurrence will be discussed.
Cases
Case 1
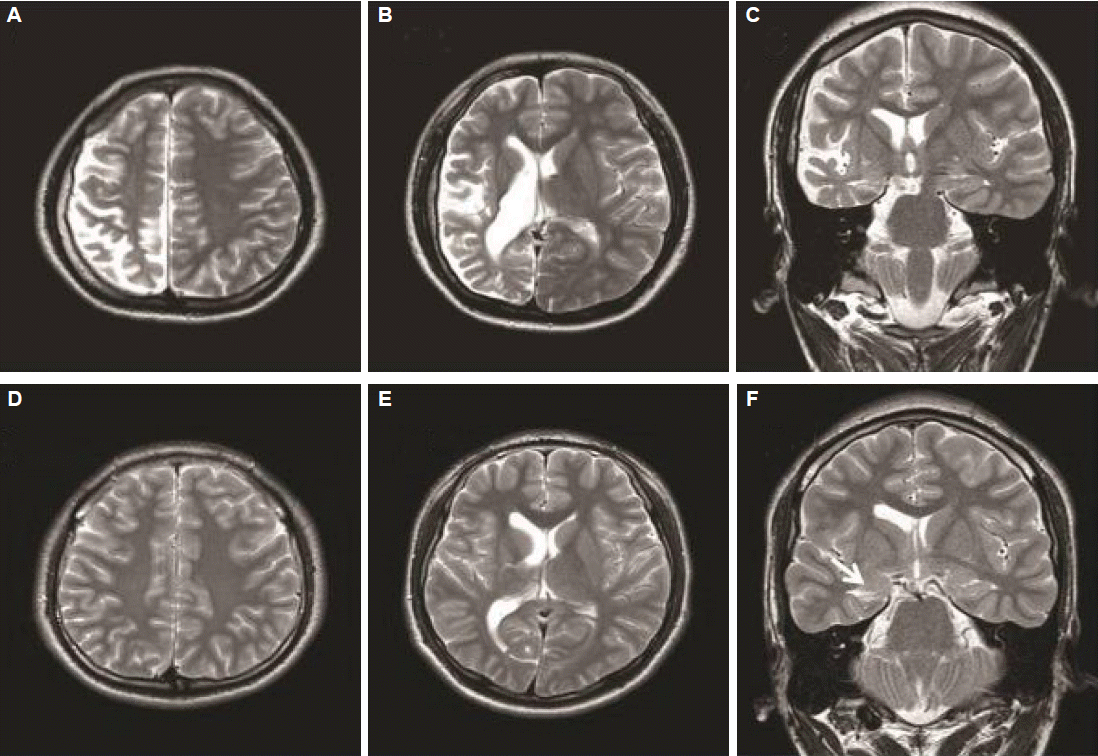
A 26-year-old man was referred for consultation about his recurrent seizures and hemiparesis. He was born by cesarian section for breech position, but in good condition. However, he had displayed developmental delay since infancy. He could express meaningful words at the age of 3, and showed a hemiplegic gait since he first started walking. The patient had experienced seizures since the age of 4. From the age of 4 to the time of referral, his seizures were yearly events regarding generalized tonic-clonic (GTC) seizure and weekly regarding aura-only. Neither febrile convulsion, nor meningoencephalitis occurred. The neurological examination revealed mild hemifacial weakness and hemiparesis on the left side. The intelligence quotient was 66. He experienced focal motor seizures in the left arm and leg, independently or prior to a GTC seizure. In some instances, he noted an auditory aura with or without being followed by a brief alteration of consciousness or secondary generalization. His brain magnetic resonance image (MRI) is demonstrated in Fig. 1A, B, and C. Repetitive electroencephalographies (EEGs) demonstrated continuous irregular theta slow activities and lower amplitude over the right hemisphere (Fig. 2A).
Case 2
The brother of case 1 was 3-year younger. He had also suffered from epilepsy since the age of 2. He was born at term without any complication, and showed normal development. Also, he had not experienced febrile seizure or meningoencephalitis. From the age of 2 to the last year before a first visit, he experienced seizures with a frequency of approximately one per year. Its semiologies varied as follows: generalized seizures, often preceded by visual or cephalic auras, or an auditory aura such as tinnitus. The generalized seizures and auditory auras had recently frequently recurred at one or two per month. Neurological examination revealed normal finding and his intelligence quotient was 96. The EEG showed continuous theta or delta activity in the right hemisphere, which was accompanied by occipital sharp waves (Fig. 2B). The brain MRI findings are shown in Fig. 1D, E and F.
Discussion
The Case 1 displayed the characteristic radiological and clinical findings that are consistent with a diagnosis of DDMS. Based on the long-term history, the stationary pattern in this patient regarding the seizure occurrence, his cognition and the hemiparesis, made Rasmussen encephalitis unlikely, which has a progressive nature of seizure and neurologic deficit, despite of the similarity of the imaging findings. The patients with Sturge-Weber syndrome usually have typical skin lesions which is also incompatible with this patient. Besides, both diseases mentioned above tend not to have skull thickening. However, diagnosing the second case as DDMS may be debatable, because case 2 did not show the definite hemispheric atrophy, skull thickening and mental retardation consistent with DDMS, like his brother’s. Furthermore, asymmetries of the ventricles have been known to be a normal variant in some population, particularly the occipital horns.3 Nonetheless, the result of EEG indicating continuous irregular slow activity over one hemisphere, hippocampal atrophy and small caudate body in the same side with that of small ventricle might support a possibility of abnormal or dysfunctional hemisphere rather than normal variant. Therefore, case 2 might be regarded as having a mild form of DDMS. Notably, the first case had just yearly seizures even though the imaging is more typical and severe than the second case, who had drug-resistant seizures and was considered as a potential candidate for surgery. Taken previous cases (Table 1) and ours together, the severity of seizure and radiological findings seem to be unrelated with each other.
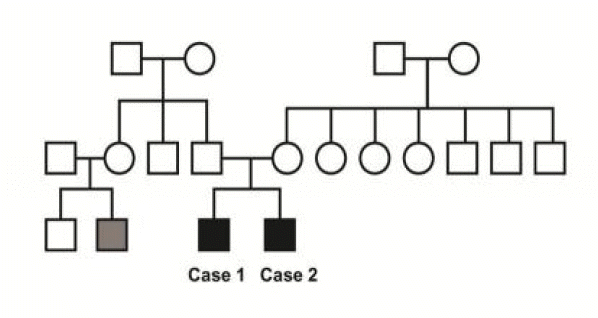
Of interest, the both patients were brothers and they had another relative suffering from epilepsy (Fig. 3). Rather than coexistence, a genetic contribution in the development of this syndrome might well be presumed. Conventionally, the congenital causes of DDMS include congenital malformation, intrauterine vascular occlusion and infection,1,4–13 but genetic cause, neither the case of DDMS in a epilepsy family, has not been previously reported. For the semiological aspect, case 1 showed several different types of seizure, which were focal motor or auditory with/without secondary generalization. Case 2 mainly showed an auditory aura. This common semiological feature was consistent with that of a previous report that documented familial temporal lobe epilepsy (TLE) with an auditory aura.14 Contrary to their similar semiology, discordant data showing occipital epileptiform discharges on EEG and abnormal medial temporal area on MRI, do not support this assumption.
Another possibility is that HS, as a common image finding between the two brothers, might be the sole familial proportion of genetic inheritance. In addition, the different degree of cerebral asymmetry could be a reflection of different seizure-induced neo-cortical change between two brothers. However, this seems unlikely because the seizure frequencies were similar in the two brothers and the onset of hemiparesis in case 1 preceded the seizure onset. A previous study of patients with cerebral hemiatrophy showed that the HS in the study subjects was strongly related with a history of febrile convulsion.15 Since there was no history of febrile convulsion in the brothers of our study, it does not appear that febrile convulsion intrudes to the explanation of our familial epilepsy case. There also might have been the coexistence of DDMS and common non-genetic TLE, and the unrevealed environmental factors occurring in this family might have affected the development of epilepsy, as is often the case of families with various diseases.
In order to confirm the possible genetic component in DDMS, a detail family history should be sought in a patient with the features of DDMS and further reports are mandatory.