Introduction
Status epilepticus (SE) is a neurological emergency that may result in serious morbidity and mortality.1 Magnetic resonance imaging (MRI) is an essential tool for determining the underlying cause of SE and can effectively demonstrate many pathophysiologic changes, such as cerebral edema, hyperperfusion and alteration of the blood-brain barrier. SE can exhibit a variety of unpredictable MRI findings that can be focal, multifocal, hemispheric or diffuse.
We report the case of a previously healthy woman who exhibited bilateral increased T2 signal in the external capsule on MRI following SE.
Case
A 28-year-old woman visited our hospital due to a sudden onset of lip smacking and unresponsiveness. Seven days prior to the hospital visit, she had developed a fever and generalized myalgia, but had not sought treatment or taken medications.
The patient had no history of epilepsy or trauma, nor was there any relevant family history. Her vital signs were within normal range except for a high fever of 37.8°C. On neurologic examination, the patient was confused, could not follow the examiner’s verbal commands and often attempted to climb out of bed. Neck stiffness was not observed.
Complete blood counts were normal. Cerebrospinal fluid was clean and contained no leukocytes, with normal glucose (73 mg/dL) and protein (41.5 mg/dL). The results of serologic analysis of blood and cerebrospinal fluid (CSF), including polymerase chain reaction for herpes viruses, were within normal range.
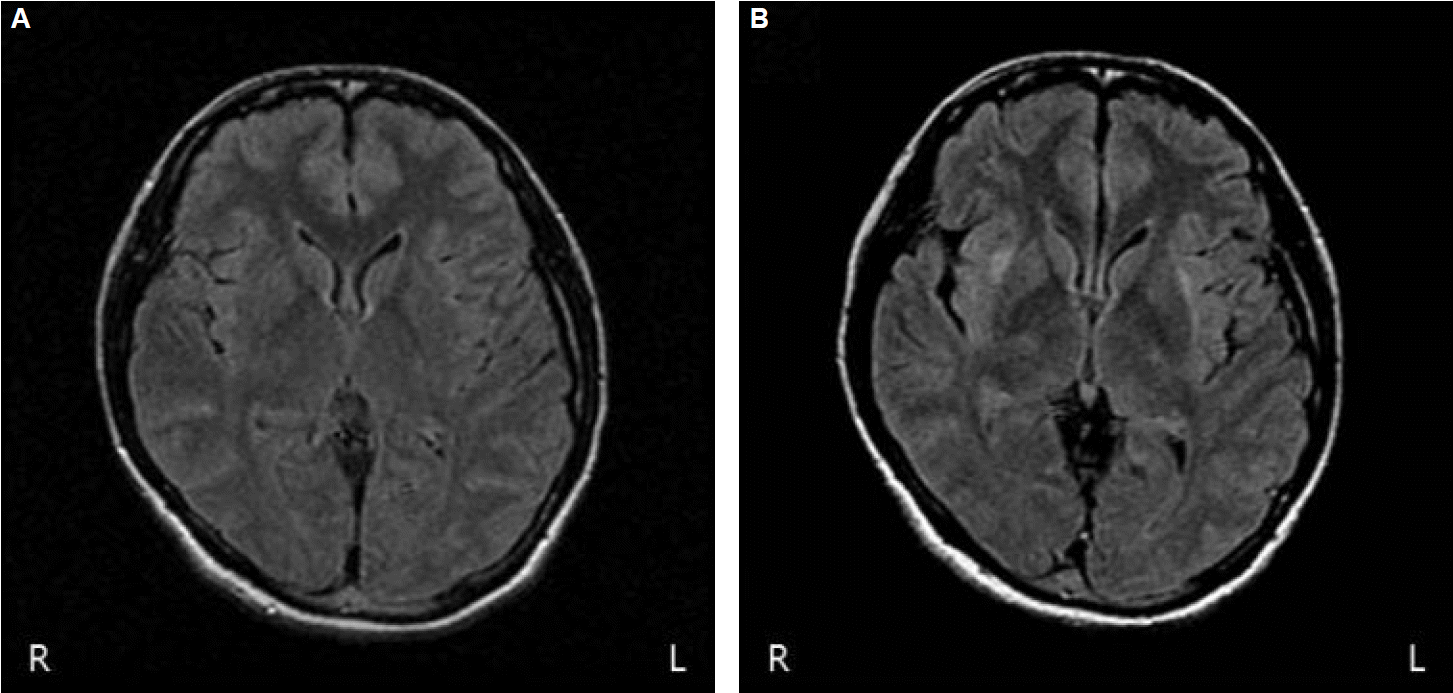
Brain computed tomography and initial brain MRI revealed no abnormalities (Fig. 1A). Diffuse slow-wave discharges were observed on electroencephalography (EEG).
Seizure did not recur after one hour of continuous infusion of 15 mg/kg of phenytoin mixed with normal saline. On the day of admission, we started acyclovir because of the clinical suspicion of herpes encephalitis.
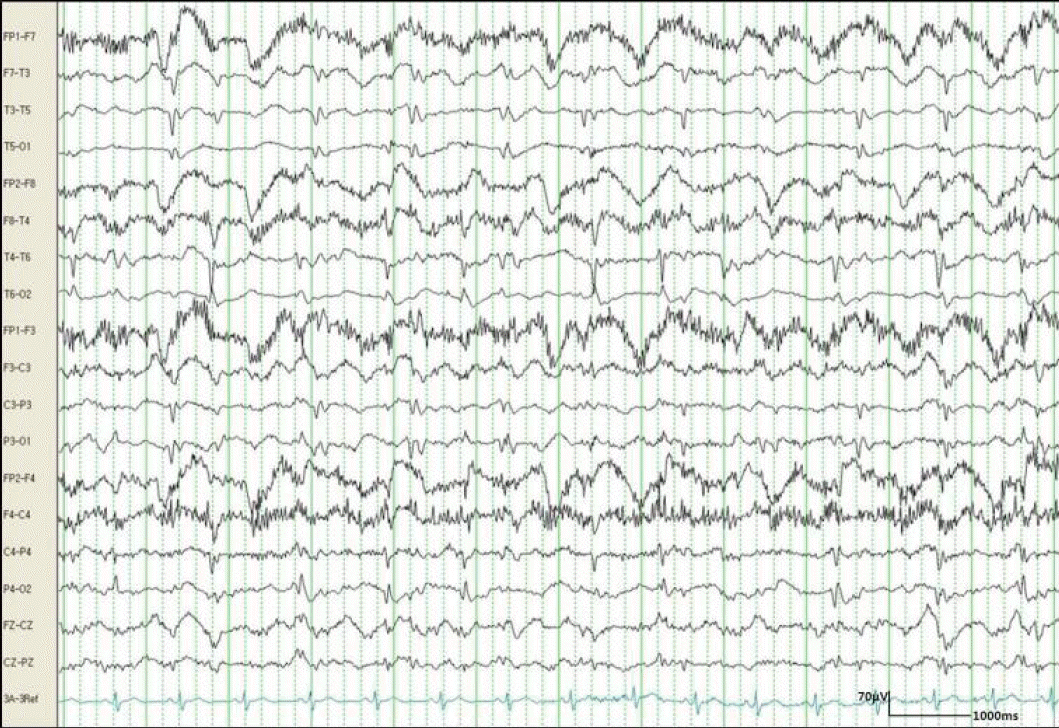
Three days after admission, the frequency of seizures gradually increased without full recovery of consciousness. EEG revealed a repetitive generalized spike and waves at 1–1.5 Hz (Fig. 2), so we administered additional antiepileptic drugs (lamotrigine and levetiracetam) and began midazolam-coma therapy.
Twenty days after admission, the patient recovered full consciousness with no further seizures. However, she had a difficulty in standing alone and walked with assistance for 2 days. She slowly responded to the questions. She complained short-term memory impairments such as a failure to recall the persons who had visited her several hours ago. There was no other focal neurologic deficit. Follow-up MRI demonstrated hyper-intensities lesions in the right claustrum and bilateral external capsular areas on T2-fluid attenuated inversion recovery (FLAIR) images (Fig. 1B). Apparent diffusion coefficient maps did not evidence decreased diffusion. The patient’s postural and cognitive symptoms resolved spontaneously without any change in medication.
Discussion
This report describes a patient with SE presenting as hyper-intensities on the T2-FLAIR image of the right claustrum and bilateral external capsule.
MRI has become the most important tool for observing neuroanatomic changes during SE. In a previous report, MRI changes due to SE were reported in 11.6% of cases.2 Various MRI changes related to SE have been previously reported; most were observed in mesial temporal structures such as the amygdala, hippocampus or cortical area, and were rarely observed in the basal ganglia, thalamus, brain stem or cerebellum.3–5 The occurrence of external capsule or claustrum lesions without other lesions has only been encountered in few case reports.5–7 MRI changes were seen not only in the areas primarily involved in seizure activity, but also in those remote from, but functionally connected to, the epileptogenic cortex. Changes in the bilateral external capsule may reflect the propagation of epileptic seizure discharges involving widespread network interactions between cortical and subcortical structures.
The pathophysiological basis for MRI changes related to SE remains unclear, but it may reflect various neuronal injuries resulting from sustained seizure activity. Prolonged seizures are known to cause neuronal injury by various mechanisms: deficiency of adenosine triphosphate leading to anaerobic metabolism and lactic acidosis; impaired ion channel and increased membrane ion permeability of the cell; release of inflammatory mediators; and possible activation of caspase pathways leading to apoptosis.4,8 The observation of abnormal MRI signal after SE likely denotes these neuronal injuries.
The consequences of structural changes related to SE have been described less frequently. It is well known that patient with hippocampal change after SE presents intractable seizure. Extratemporal changes in the basal ganglia (BG), thalamus and cerebellum have not been associated with sequelae related to structural lesions.5–7 One observation of focal cortical change following focal motor SE indicated residual neurologic deficits.9
In this report, transient postural instability and cognitive impairment were clearly observed after SE. As the external capsule is a route for cholinergic fibers from the basal forebrain to the cerebral cortex, cognitive dysfunction might be related to these fibers. In addition, external capsule is thought to contain corticostriatal fibers connecting the cerebral cortex and the striatum. Involvement of these fibers might be related to postural instability.10
Although the exact cause or mechanism of MRI change following SE has not yet been identified, it is a useful tool for better understanding of the pathophysiology of neurologic symptoms related to SE.