Introduction
Status epilepticus (SE) is a medical emergency. The general clinical course of a typical convulsive status can be categorized as: early, established, refractory and super-refractory. SE that continues or recurs 24 hours or more after the onset of anesthesia, including those cases in which SE recurs on the reduction or withdrawal of anesthesia is known as super-refractory status epilepticus.1 While treatment guidelines exist for established and refractory status epilepticus, the management of super-refractory status epilepticus is not well established. We present here a case of super-refractory status epilepticus treated with doses of anesthetic medications beyond the recommended guidelines with a good clinical outcome.
Case
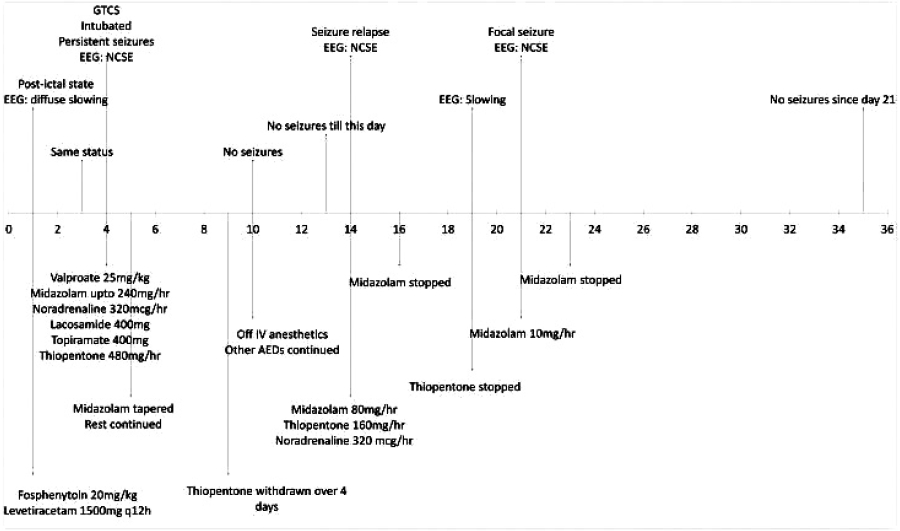
A 28-year old female, a known hypertensive for 2 years, had 2 episodes of generalized tonic-clonic seizures (GTCS) following 2 days of fever. She was conscious in the interictal period. She had no prior history of seizures. She was managed initially at another hospital where she was given a loading dose of 3 g of levetiracetam. The next day, she was brought to our emergency department in a drowsy state following 2 more GTCS. On examination, she did not have any focal neurological deficits. She was given a loading dose of fosphenytoin (20 mg/kg phenytoin equivalent), while levetiracetam was continued at 1,500 mg q12h. MRI brain with and without gadolinium contrast revealed no abnormalities. Routine blood chemistries and hemogram were normal. Creatine phosphokinase was normal. Electroencephalogram (EEG) showed diffuse slowing without any interictal epileptiform discharges. The differential diagnosis was viral vs autoimmune encephalitis. At the time of admission, cerebrospinal fluid (CSF) analysis was normal (glucose 66 mg/dL with blood glucose 118 mg/dL, protein 48 mg/dL, 2 cells, both lymphocytes). CSF polymerase chain reactions (herpes simplex virus and tuberculosis) were negative. Her thyroid profile and anti-thyroid peroxidase antibody titres were within normal limits. She was treated with acyclovir empirically.
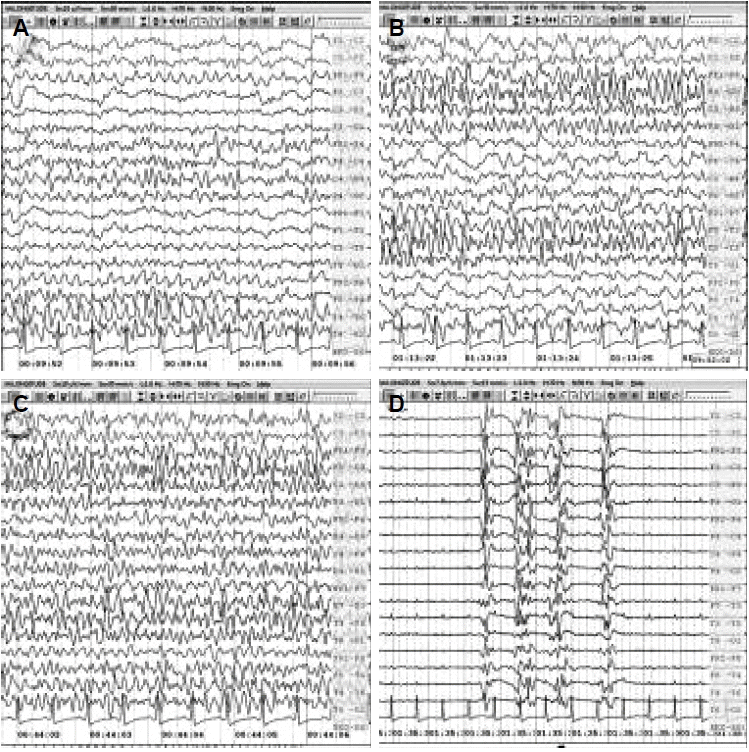
She regained consciousness and was stable for the next 3 days. On the 4th day, she developed one episode of GTCS. Clobazam was added at a dose of 10 mg qhs. Following this episode, she developed recurrent focal seizures originating from the right as well as left temporal regions with worsening of sensorium (Fig. 1A). She was intubated and placed on mechanical ventilator support. A loading dose of sodium valproate (40 mg/kg body weight) was given. For the next 2 hours, no clinical or electrographic seizures were recorded.
After 2 hours, there was a recurrence of focal seizures with mild facial twitching, amounting to non-convulsive status epilepticus (NCSE, Fig. 1B). A loading dose of midazolam (0.2 mg/kg) was followed by maintenance infusion which was gradually increased to 3 mg/kg/hr (body weight 80 kg, total dose was 240 mg/hr). At this dose of midazolam infusion, she developed hypotension with blood pressure of 80/60 mmHg. At this stage, noradrenaline infusion was initiated and the dose was titrated (320 mcg/hour) with concurrent decrease in the rate of infusion of midazolam (decreased upto 80 mg/hr). She had relapse of seizures in the form of recurrent electrographic seizures. Lacosamide (400 mg loading, then 200 mg q12h) and topiramate (400 mg loading, then 200 mg q12h) were added to the regimen. Due to continuation of NCSE, thiopentone sodium infusion was initiated and gradually increased. At a dose of 6 mg/kg/hr (total dose 480 mg/hr), burst suppression was achieved. At this point, her hemogram, renal and hepatic parameters were within normal limits. After 5 days of IV methylprednisolone (IVMP), she was given 5 days of IV immunoglobulin (0.4 g/kg/day).
Evaluation for autoimmune state, including antibodies against N-methyl D-aspartate receptor and voltage-gated potassium channel as well as for systemic vasculitis was normal. As the CSF parameters, and investigations for systemic autoimmune conditions were normal, we postulated a probable diagnosis of viral vs autoimmune encephalitis. After 12 hours of burst suppression, gradual withdrawal of midazolam and thiopentone was started. Midazolam was completely stopped and thiopentone was reduced to 40 mg/hr without seizure recurrence, following which it was completely stopped.
However, she developed recurrent electrographic seizures again. The second cycle of midazolam and thiopentone was started with doses of 80 mg/hr and 160 mg/hr respectively. Vasopressor support was added for hypotension. With these doses, control of seizures was achieved and they were gradually tapered off over the next 4 days. Her sensorium improved and she was able to follow simple commands.
After 5 days, she developed a focal seizure with worsening of sensorium. EEG showed frequent electrographic seizures (Fig. 1C). Midazolam was restarted at 10 mg/hr and control was achieved at a dose of 50 mg/hr. MRI Brain and CSF analysis were repeated but did not show any abnormalities. Midazolam was tapered and stopped over the next 2 days after seizure control (Fig. 1D). Acyclovir was continued for a total of 14 days.
She gradually improved and was weaned off the ventilator. There was no further seizure recurrence. She had developed mild proximal weakness of all limbs which was diagnosed as critical illness myoneuropathy. Cognitive assessment showed severe attentional deficits.
On discharge, she had mild proximal weakness, attention and occasional recent memory deficits. She continued to improve during the follow-up period. At the last visit, she had no residual weakness but had mild attention and recent memory deficits. She was able to take care of herself and her family independently.
The major events of the case are summarized in a timeline in figure 2.
Discussion
About 15% of all status epilepticus progress to super-refractory status epilepticus. The causes postulated include excitotoxicity, inhibitory neurotransmitter dysfunction, energy failure, inflammatory processes and network reorganization. The aim of treatment is to control seizures and prevent propagation of damage.2 However, the treatment of super-refractory status epilepticus is difficult and is hampered by the lack of established guidelines.
The best treatment, of course, is treatment of the cause. For control of seizures, various methods have been proposed. These include anesthetic agents (thiopentone, midazolam, propofol, ketamine and inhalation anesthetics), newer antiepileptics, magnesium, pyridoxine, immunotherapy, ketogenic diet, hypothermia, neuromodulation and surgery.2
The doses for midazolam and thiopentone for adults, as used in available literature, are up to 0.4 mg/kg/hr and 5 mg/kg/hr respectively.2 In our case, we used midazolam up to 3 mg/kg/hr and thiopentone up to 6 mg/kg/hr. Even at these doses, hypotension was transient and there were no other systemic complications. We postulate that this case is unique in the sense that the patient had only transient hypotension with anesthetic medications and the good clinical outcome is related to appropriate aggressive medical care in a young female.
The outcome of patients with refractory and super-refractory status epilepticus (SRSE) is variable.3,4 As we gain more information, the prognosis of super-refractory status epilepticus will improve.
In the absence of established guidelines, the treatment of super-refractory status epilepticus needs to be individualized. Aggressive treatment, even with doses beyond those conventionally used, can result in a good functional outcome with minimal complications.