Introduction
Aphasic status epilepticus (ASE) is a very rare clinical manifestation. Different from nonconvulsive status epilepticus (NCSE), cognitive dysfunctions such as decreased responsiveness and awareness are absent in ASE.1–3 Usually, ASE is associated with structural brain lesions such as brain tumor, trauma, and stroke.1,2 Metabolic disturbance without structural brain lesions is scarcely associated with ASE.2,4,5
We herein describe the case of a patient with ASE associated with acute aggravation of uremia and discuss the mechanisms and points of differentiation from other disorders.
Case
A 76-year-old female was brought to the hospital by her family following 1 week of language disturbance. The symptom developed suddenly and progressed slowly. She recurrently showed speech arrest ranging from a few minutes to an hour several times a day. She had been suffering from chronic kidney disease and had received peritoneal dialysis for 12 years. She neglected receiving dialysis 3 weeks prior to hospitalization because of her depressed mood. Before visiting our hospital, she visited another hospital and was diagnosed with functional aphasia due to depression and administered placebo followed by antidepressant, but no clinical improvement was observed.
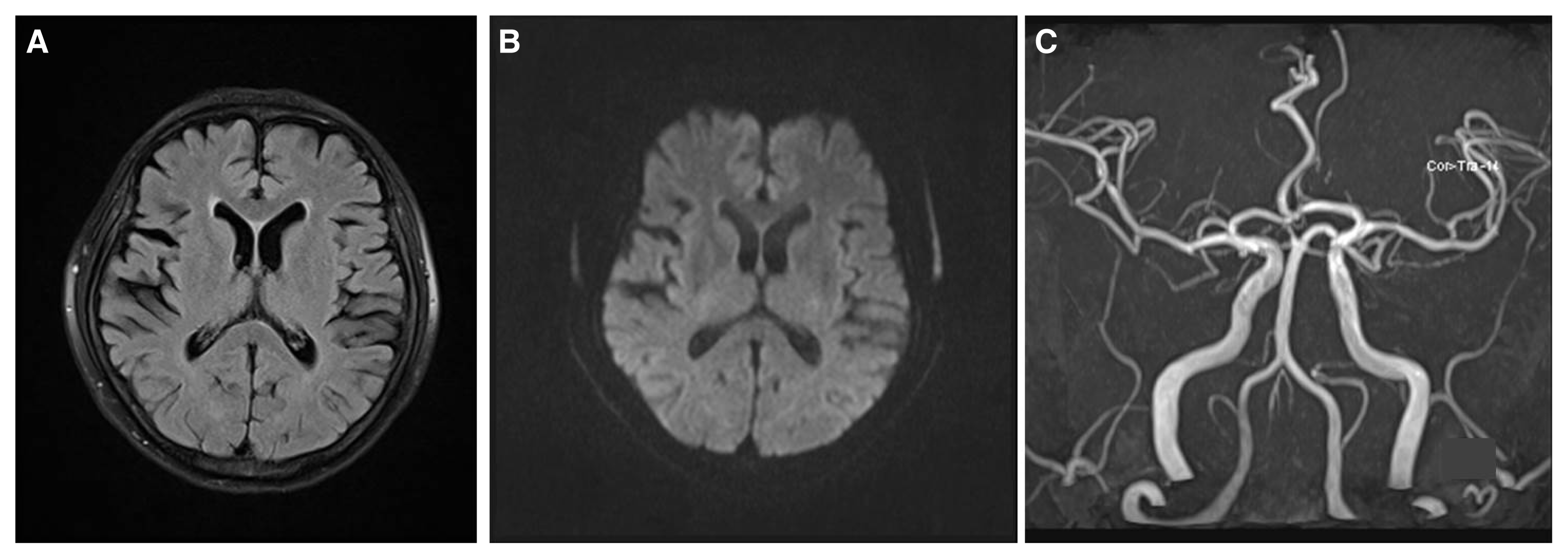
Upon neurological examination, she was alert and no focal neurologic deficit other than aphasia was observed. She could walk unassisted. She showed recurrent motor aphasia, and could obey simple commands but could not speak for about 10 minutes. The aphasic symptom then disappeared and she was able to speak for a few minutes. No abnormal behaviors, such as confusion, motionless staring, automatisms, or wandering were observed. We presented her with three figures (a hammer, a coin, and a key) and asked her to remember the items. Routine laboratory testing showed that the value of serum blood urea nitrogen and creatinine were 62.7 mg/dL (7.8–22.0 mg/dL) and 12.4 mg/dL (0.6–1.2 mg/dL), respectively. They were increased compared with the respective values of 56.7 mg/dL and 9.7 mg/dL measured 1 month before. Her hemoglobin level was 6.8 g/dL (12.0–16.0 g/dL), her glucose level was 79 mg/dL (70–110 mg/dL), her sodium level was 141 mMol/L (135–145 mMol/L), her potassium level was 7.3 mMol/L (3.5–5.3 mMol/L), and her ionized calcium level was 0.73 mMol/L (1.13–1.32 mMol/L). Serum osmolality was measured as being 336 mOsm/Kg (289–308 mOsm/Kg). Brain magnetic resonance imaging and angiogram, including diffusion weighted imaging, revealed no abnormality (Fig. 1).
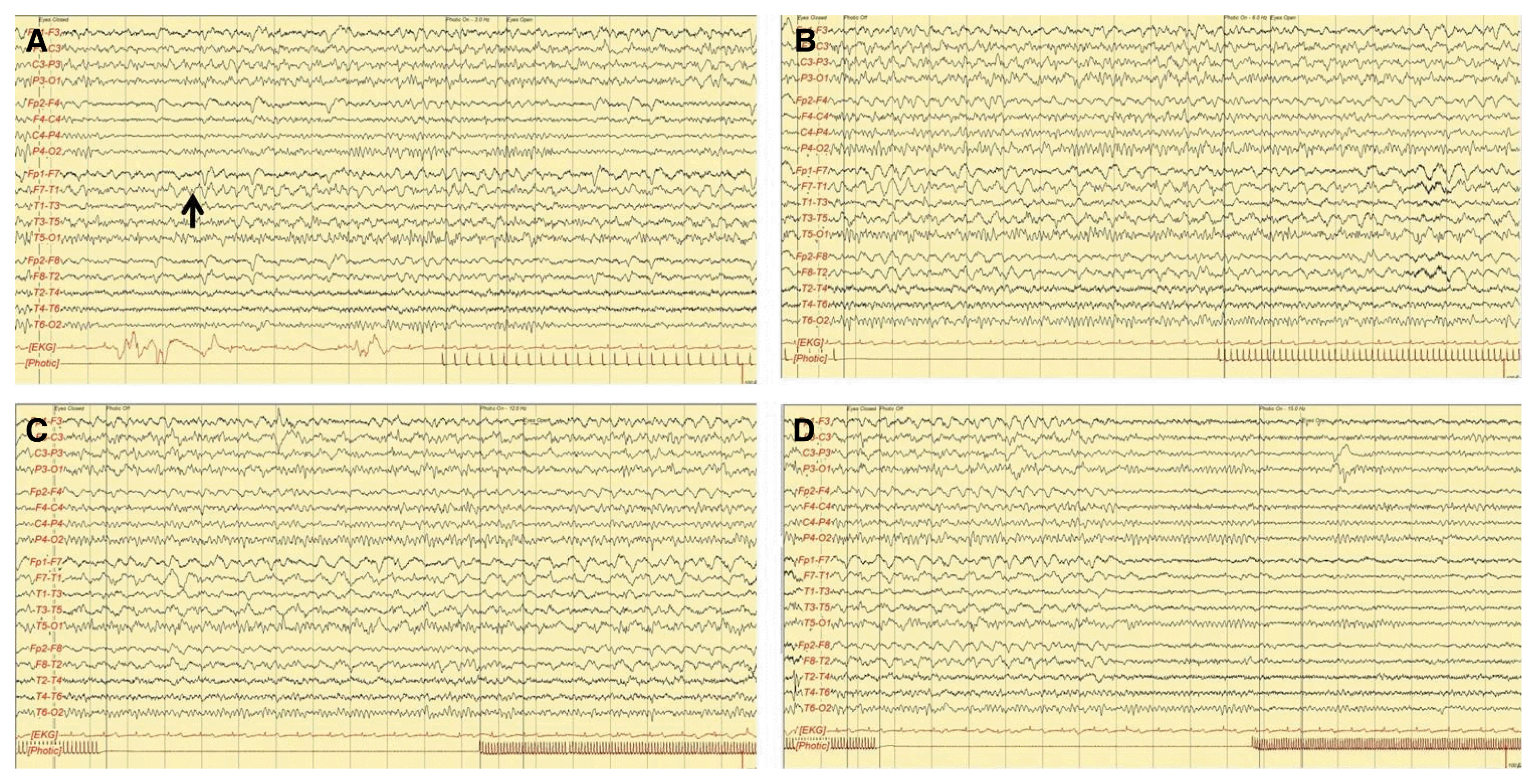
On electroencephalography (EEG), ictal rhythmic discharges were observed in the left frontotemporal area over approximately 1 minute, and repeated four to five times per hour (Fig. 2). After administering 1 g of intravenous valproic acid, she became fluent and the ictal EEG discharges disappeared. She could remember the events of the aphasic period. She could remember two of the previously presented figures (a hammer and a key). She was transferred to the nephrology department for the treatment of uremia and hyperkalemia. Administration of valproate continued with maintenance dose of 1,000 mg/day for 2 weeks. We decided to discontinue medication because the symptom did not recur. She has remained symptom-free for 6 months after correction of the metabolic disturbances.
Discussion
Aphasia is usually caused by structural brain lesions, such as stroke or tumors affecting the language network, and is evident on neuro-imaging in almost cases. However, unlike our patient, the symptom is usually persistent in those disorders, with no symptom-free periods. NCSE should be considered in patients with recurrent impaired speech, but altered responsiveness and awareness is more prominent in patients with NCSE than ASE.1–3 Our patient was alert and could remember the events of the language disturbance. Memory function was retained during the ictal period. These findings are compatible with ASE.
Occasionally, ASE is misdiagnosed as functional aphasia with psychiatric disturbance. A previous report indicated that ASE was diagnosed after 10 years of antipsychotic medication.6 Without clinical suspicion, diagnosis can be delayed and lead to irreversible neurologic sequalae.
According to previous case reports, ASE is associated with symptomatic brain lesions. Nearly all patients have lesions in the vicinity of the language cortex.1,2 Only in a few case reports, the patients had ASE in the setting of metabolic disturbance, and all of them were associated with nonketotic hyperglycemia (NKH).2,4,5 Two of these cases showed focal cerebral hypoperfusion on single-photon emission computed tomography in the interictal period.4,5 The exact mechanisms of focal seizures in NKH are uncertain. One proposed mechanism is that dehydration may cause acute focal reduction of cerebral blood flow and excitation of the cortical neurons in the area, as well as hyperglycemia and hyperosmolality.4 Similar to patients with NKH, hyperosmolality and dehydration may have played a role in producing seizures in our patient. Another possible mechanism is direct uremic toxicity on cerebral neurons. Uremic toxins such as guanidine compounds can enhance cortical excitation;7 they are antagonistic to GABAergic receptors and agonistic to N-methyl-D-aspartate receptor (NMDA receptor), and cause seizures in patients with renal failure. Other laboratory abnormalities of our patient, such as hypocalcemia and hyperkalemia, may have been related to the seizures.
In our case, other metabolic abnormalities, such as hyperkalemia, severe anemia, and hypocalcemia, were also observed. However, the potassium level was not so high as to cause neurologic symptoms. Hemoglobin and calcium levels were significantly low, but were chronically lowered in this case. These laboratory abnormalities are less likely to have caused seizures.
In conclusion, this is the first case report of ASE in a patient with acute aggravation of uremia. ASE may be caused by uremia without a structural brain lesion. EEG allowed for early detection of seizure activity. Early detection and treatment with short-term administration of an antiepileptic drug and correction of uremia can prevent further ASE and permanent neurological disability.