Introduction
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is an uncommon neurocutaneous disorder, with an incidence of 0.7 per 100,000 live births.1,2 IP is a genetic disorder resulting from mutations in the gene encoding inhibitor of kappa-B kinase gamma (IKBKG), a protein that activates nuclear factor kappa B (NF-κB) essential modulator (NEMO).3 Because of its X-linked dominant inheritance pattern, IP predominantly (i.e., in 95% of cases) affects female infants.4 Clinically, IP usually presents as skin lesions, but may also include ocular, dental, and central nervous system (CNS) abnormalities. Neurological manifestations associated with IP may include seizure, epilepsy, microcephaly, and encephalomyelitis. Among the manifestations, neonatal seizures are most common. Rarely, IP may appear later in life.1,2,5
This report describes a 6-month-old female with IP who exhibited late-onset status epilepticus following respiratory syncytial virus (RSV) infection.
Case
A 6-month-old female patient was referred to our hospital following 4 days of persistent fever, productive cough, multiple and prolonged generalized tonic-clonic seizures accompanied by eyeball deviation with loss of consciousness lasting for more than 30 minutes, and hyperpigmented skin lesions. The child was born via spontaneous vaginal delivery at a gestational age of 36 weeks and 4 days and had a birth weight of 2,600 g. Immediately after birth, she experienced blistering skin lesions, but neurologic examination and brain sonography showed no remarkable findings. The vesicular skin lesions gradually transformed into hyperpigmented patches.
Pedigree analysis revealed that her mother and maternal grandmother had vesicular skin lesions at birth and characteristic teeth, and that her mother had retinal problems. Vital signs showed a blood pressure of 95/60 mmHg, a pulse rate of 120 beats/minute, a respiratory rate of 24 breaths/minute, and a body temperature of 38.2°C. Her body weight, height, and head circumference were all within the normal range for her age.
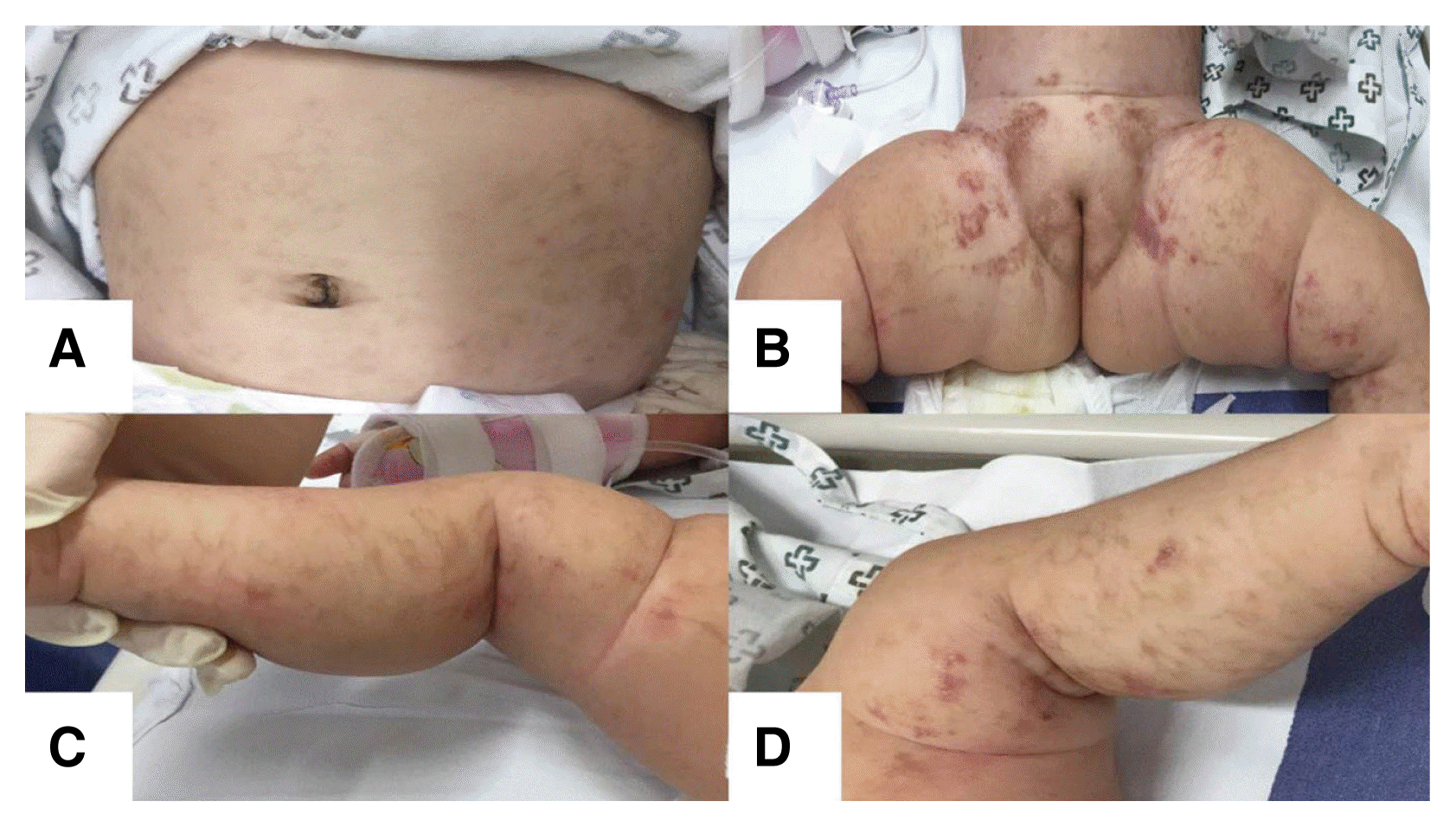
Physical examination showed hyperpigmented linear patches on her abdomen, trunk, and lower extremities (Fig. 1). Auscultation of the lungs revealed a wheezy breath sound. She had normal tone in all extremities without pathologic reflexes. Results of the Korean developmental screening test for infants and children showed no developmental delays in gross or fine motor skills, social development, language, or cognitive-adaptive function. Other assessments, including those of her dental, skeletal, and ophthalmic systems, revealed nothing abnormal.
Laboratory findings indicated no abnormalities in her blood. Cerebrospinal fluid (CSF) examination showed 2 white blood cells/mm3, a glucose concentration of 68.5 mg/dL, and a protein concentration level of 36.2 mg/dL. The CSF culture was sterile. Respiratory virus real-time polymerase chain reaction (PCR) from nasal swab showed a positive reaction for RSV group B.
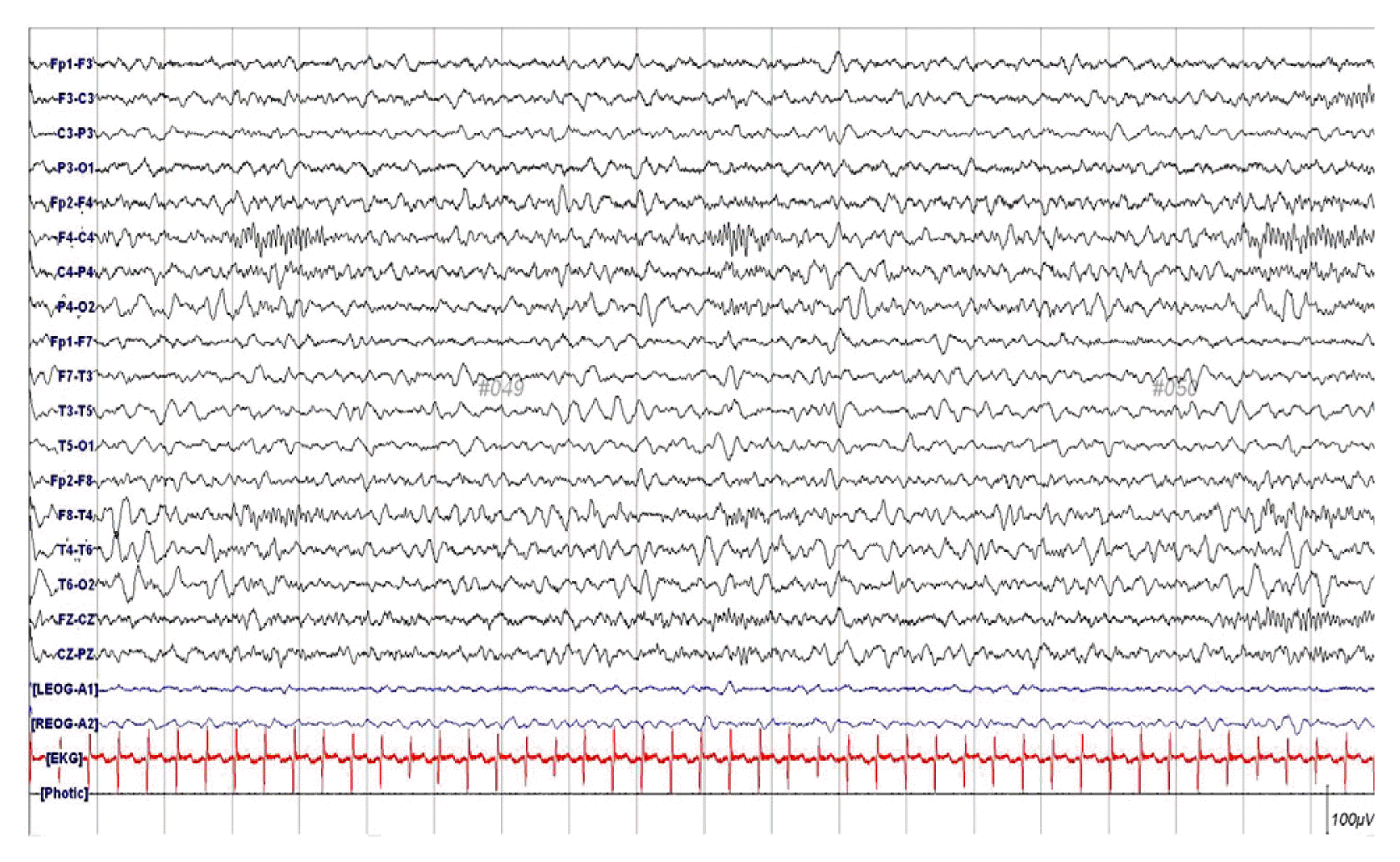
Electroencephalography showed asymmetric voltage attenuation on her left hemisphere (Fig. 2). Magnetic resonance imaging (MRI) revealed multifocal T1 low and T2 high signal changes in both her cerebral cortex and adjacent white matter. Diffusion-weighted imaging (DWI) showed multifocal high signal intensity and susceptibility-weighted imaging (SWI) showed punctate low signal foci. These lesions manifested as areas of patchy enhancement on post-contrast scans (Fig. 3).
Molecular genetic testing was performed using long range PCR and direct sequencing to identify IKBKG mutations. An 11.7 kb deletion on chromosome Xq28, encompassing exons 4–10 of the IKBKG gene, was found.
She was treated with intravenous diazepam (0.2 mg/kg), lorazepam (0.1 mg/kg), and fosphenytoin (20 mg/kg) to halt her recurrent seizures. Following discharge, she was treated with oral anticonvulsants to reduce the persistence and recurrence of seizure. To date, she has shown appropriate developmental progress and has had no further seizures.
Discussion
IP is characterized by abnormalities of the ectodermal tissue, including the skin, CNS, teeth, and eyes. Mutations of the IKBKG gene, located at chromosome Xq28, are responsible for IP. Approximately 80% of patients with IP have an 11.7 kb deletion of exons 4 to 10 in the IKBKG gene.6
Dermatological features of IP may arise at birth and usually occur within the first 2 weeks of life. The cutaneous signs of IP typically progress over time in four stages, consisting of linearly arranged erythematous vesicles (stage 1), hyperkeratotic or verrucous lesions (stage 2), hyperpigmented streaks along Blaschko’s lines (stage 3), and hypopigmented whorls (stage 4).3 Our patient presented with stage 1 cutaneous lesions immediately after birth and stage 3 skin manifestations at 6 months.
Neurological manifestations related to IP vary and may include a single seizure, ischemic stroke, cerebellar ataxia, epilepsy, and psychomotor delay. These neurologic features are observed during the first month in 66.9% of patients.5 Acute neurological signs in the newborn period mimicking encephalitis occur mainly within 4 days of birth and include feeding intolerance, lethargy, and seizure-like activity.2 These seizures are generally characterized by clusters of focal clonic seizures with loss of consciousness for a short duration (typically 5 minutes), which was unlike our case.7 In most cases, these neurological manifestations are associated with cerebrovascular damage, which has been documented by brain imaging studies.8
Our patient experienced status epilepticus following RSV infection at age 6 months. CSF examination and MRI findings were not compatible with RSV encephalitis, but rather with typical IP. Elevated levels of interleukin-6 and tumor necrosis factor-alpha (TNF-α) during the first year of life may be among the inflammatory causes of IP.9 One patient experienced recurrent inflammatory skin lesions that may have been triggered by a febrile episode, suggesting that stimuli, such as proinflammatory cytokines (e.g., TNF-α) and infectious viruses enhance apoptosis in IP patients with mutated IKBKG genes.10 IKBKG activates NEMO, which is required to activate NF-κB, a factor that regulates inflammatory responses and protects against apoptosis.7 Therefore, mutations in the IKBKG gene can inactivate the NF-κB pathway. DWI has shown multifocal high signal intensity in the deep or subcortical white matter of IP patients, whereas SWI has identified punctate low signal foci, which tends to be secondary to inflammation and hemorrhagic pathology.2 High-dose cortico-steroid therapy was found to be effective in managing the neonatal dermopathy and encephalopathy associated with IP, resulting in improvements in skin lesions and a dramatic reduction in seizure activity by decreasing TNF-α activity.6 Thus, we suspect that inflammatory responses following RSV infection may have triggered neurologic manifestations in our patient, though we did not treat her with an anti-inflammatory agent.
Prognosis depends on the severity of clinical manifestations, especially in patients with neurological features.8 The presence of neurologic features in the newborn period is considered as a poor prognostic factor.4 Thus, neuroimaging should be performed to detect latent neurological involvement in patients with skin lesions characteristic of IP, thereby avoiding sequelae.
Our patient, who had characteristic skin lesions of neonatal IP, experienced late-onset status epilepticus. Therefore, patients who experience status epilepticus should be meticulously examined for skin lesions to determine whether they have neurocutaneous syndromes such as IP. As well, a long-term multidisciplinary approach is essential for the prediction of the occurrence of other potential complications later in life and better management of IP.