Introduction
Epilepsy is a common neurological disorder estimated to affect 65 million people around the world.1 Genetic generalized epilepsy, also known as idiopathic generalized epilepsy, constitutes one-fourth of all epilepsies.2 Over the past decade, there was a huge evolution in the technology of gene sequencing, which has made the gene tests more conveniently available. Therefore, the way of thinking about the etiologies of epilepsy, its diagnosis, and management has changed.3
Cerebral metabolism is primarily dependent on glucose for which a facilitated diffusion by glucose transporter protein 1 (GLUT1) across the blood-brain barrier is crucial. This GLUT1 is encoded by the SLC2A1 gene.4 Mutations on SLC2A1 will lead to a variety of symptoms known as GLUT1 deficiency syndrome.5 Seizures occur in around 90% of GLUT1 deficiency symptoms, which could be early-onset absence epilepsy, myoclonic-astatic epilepsy, or focal seizures. Characteristically, the presence of the focal seizure will categorize the condition as GLUT1 encephalopathy.6 In this article, we report a novel intronic variant in the SLC2A1 gene in a Saudi patient with myoclonic epilepsy. We also report a new clinical phenotype where the patient has pure myoclonic epilepsy with no focal, absence, or atonic seizures and normal developmental and cognitive functions that started in childhood rather than infancy.
Case Report
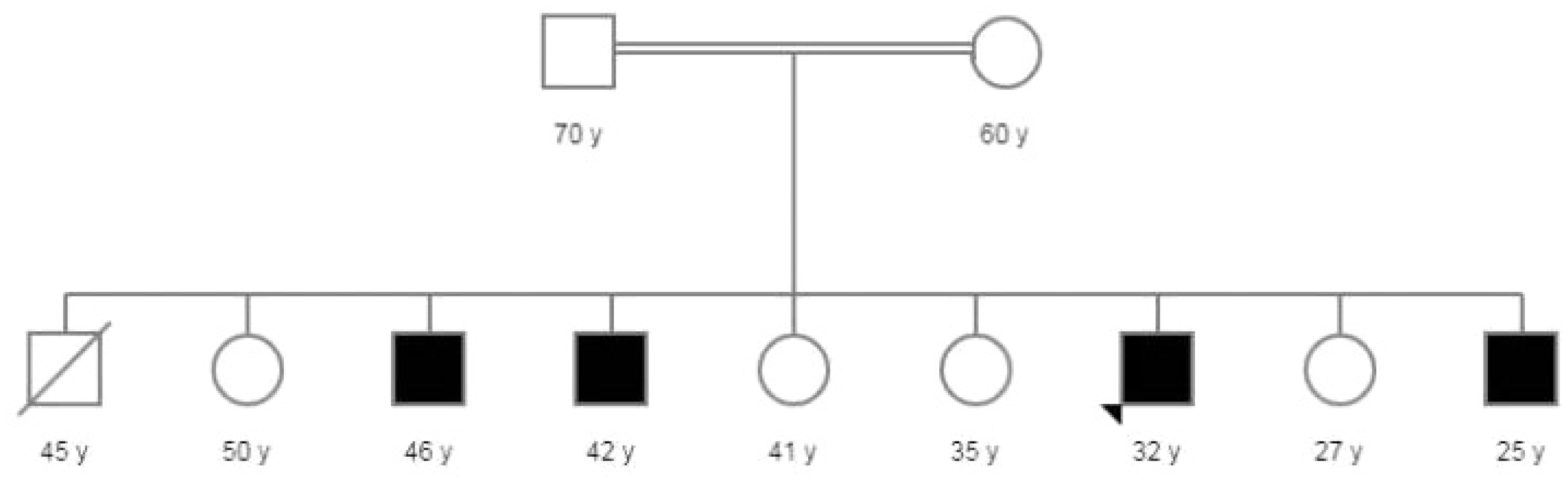
A 32-year-old male, a product of full-term pregnancy, normal uncomplicated vaginal delivery, and normal developmental milestones. He was previously healthy until the age of 10 years when his family noticed frequent myoclonic jerks involving his face, arms, and the whole body. The family sought medical advice only when he developed the first generalized tonic-clonic seizure. His past medical history was unremarkable for any neurological, metabolic, or surgical conditions. His growth, development, and school performance were normal up to the age of 10 when he dropped school due to frequent seizures. Although his seizures were so frequent, they did not interfere with the activities of daily living. His parents are consanguineous of first degree with a similar condition in four of his brothers (Fig. 1). The physical examination showed normal general examination including vital signs, skin, and no organomegaly. Higher mental functions showed a high intelligence quotient, but his speech was interrupted frequently by myoclonic jerks. Cranial nerve examination from II to VII, motor, sensory, cerebellar, and gait examination were all normal apart from brisk reflexes, which were symmetrical bilaterally. He had a brain magnetic resonance imaging (MRI), which was unremarkable. He also had an electroencephalogram, which showed epileptiform discharges in the form of polyspikes and myoclonic jerks, consistent with the diagnosis of generalized epilepsy. Whole-exome sequencing was performed and identified a heterozygous c.1278+12delC variant in SLC2A1, which is inherited in an autosomal dominant manner. The intronic variant has been identified in four heterozygous individuals in the Genome Aggregation Database (gnomAD, n>120,000 exomes and >15,000 genomes). This variant is not predicted to affect splicing by the Alamut Visual splicing prediction software (SSF, MaxEnt, NNSPLICE, GeneSplicer, and HSF). Cerebrospinal fluid (CSF) analysis showed hypoglycorrhachia (45.5% when compared to serum glucose level) with normal cytology, microbiology, and chemistry. He was treated with several anti-epileptic drugs, and currently, he is on valproic acid 1,000 mg twice daily, levetiracetam 1,500 mg twice daily, and clonazepam 0.5 mg once daily. The ketogenic diet was tried for a period of 6 months, but unfortunately, he did not respond to such therapy.
Discussion
Cerebral metabolism is totally dependent on glucose as a source of energy in the presence of glucose transporter to facilitate the transport of glucose across the blood-brain barrier into astrocytes and neurons. Several glucose transporters have been identified, among which GLUT1 has been studied most extensively.7 This important glucose transporter protein is a fundamental vehicle that facilitates glucose entry into the brain. It is encoded by the SLC2A1 gene, which is located on chromosome 1p34.2 that contains ten exons and spans 35 kb. Deficiency or abnormal function of this important transporter protein leads to neurological dysfunction with subsequent development of disease status.8 The first case report of GLUT1 deficiency syndrome was published in 1991 by De Vivo et al.9 who described two children with seizures, delayed development, and persistent hypoglycorrhachia. Both patients responded dramatically to treatment with a ketogenic diet. In 1998, Seidner and his coworkers10 published the first mutation in SLC2A1 and the symptoms of GLUT1 deficiency syndrome. By 2018, more than 300 cases were reported over the world about GLUT1 deficiency syndrome due to SLC2A1 mutation.5
The cerebral metabolic rate for glucose is low during fetal development with a linear increase after birth. It peaks around the age of 3 years and remains high for the remainder of the first decade of life. It then declines gradually during the second decade of life and plateau to the rate of glucose utilization seen in early adulthood. This might explain the onset of disease during infancy and early childhood.11 This is different from our patient who presented in the second decade of life, which might be explained by the role of other glucose transporter proteins during different stages and decades of life. This hypothesis has to be confirmed through further research.
The SLC2A1 gene (OMIM# 138140) on chromosome 1p34.2 encodes a major glucose transporter in the brain, placenta, and erythrocytes. Pathogenic variants in this gene have been identified in patients with GLUT1 deficiency syndrome, a continuum of phenotypes with overlapping features. GLUT1 deficiency syndrome is typically inherited in an autosomal dominant manner, although autosomal recessive inheritance has been reported in rare instances. It has been estimated that 90% of individuals with autosomal dominant GLUT1 deficiency syndrome have the disorder as the result of de novo pathogenic variant, and 10% have an affected parent.5
The classic GLUT1 deficiency syndrome is characterized by infantile-onset seizure, delayed neurological development, acquired microcephaly, and complex movement disorders. Several seizure types may occur including generalized tonic-clonic, focal, myoclonic, atypical absence, and atonic seizures.12 The complex movement disorder is characterized by ataxia, dystonia, and chorea, which could be paroxysmal or continuous. The severity of these abnormal movements may be influenced by fasting, fever, or infection. Patients with milder phenotypes may present only one or two of the three classical neurological symptom domains (seizures, movement disorders, and cognitive/behavioral disturbance). In addition to the classic phenotype, these phenotypes include dystonia 9, dystonia 18, atypical childhood absence epilepsy, myoclonic-astatic epilepsy, and paroxysmal non-epileptic findings such as intermittent ataxia, choreoathetosis, dystonia, and alternating hemiplegia.5
Neuroimaging of the brain including computed tomography scan and MRI are usually normal in appearance in patients with GLUT1 deficiency syndrome. In a few cases, minor or non-specific changes may be observed including enlarged inner and outer CSF spaces, slight brain atrophy, or mild delayed myelination. In contrast to neuroimaging, cerebral fluoro-deoxy-glucose positron emission tomography (FDG-PET) could be of great help to the diagnosis of GLUT1 deficiency syndrome. It reveals a huge decrease in glucose uptake in the cerebral cortex (especially the mesial temporal regions), cerebellum, and thalami with relatively preserved basal ganglia metabolism. This distinctive appearance starts in early infancy and persists into adulthood regardless of disease severity or treatment with ketogenic diet.13
The electroencephalogram (EEG) findings in patients with GLUT1 deficiency syndrome are variable and complex. The EEG may show normal activity or encompass focal, generalized, or multifocal abnormality with the same patient at different times. In infancy, focal epileptiform discharges or slowing are more frequent as compared to childhood and adolescents, where the most common EEG abnormalities are generalized 2.5- to 4-Hz spike- or polyspike-wave discharges. In the carbohydrate-responsive phenotype, the fasting EEG is usually abnormal with improvement after glucose infusion or carbohydrate intake. This situational improvement (fasting vs. postprandial) indicates reversible brain energy impairment caused by the GLUT1 defect.14 Unfortunately, in our patient, this comparison was not made.
Lumbar puncture is an important investigation for the diagnosis of GLUT1 deficiency syndrome. It usually demonstrates hypoglycorrhachia in the setting of normal serum glucose indicating impaired glucose transport into the brain. It should be performed after 4–6 hours of fasting to achieve a metabolic steady-state with simultaneous determination of serum glucose concentration.15 In our patient, CSF analysis showed hypoglycorrhachia (45.5% when compared to serum glucose level).
GLUT1 deficiency syndrome is classified as a treatable disorder with a ketogenic diet being the treatment of choice.16 The ketogenic diet is characterized by low-carbohydrate, low-protein, and high-fat diet mimicking the metabolic state of fasting. The ketogenic diet relies on ketone production through exogenous rather than body fat, thus maintaining ketosis without weight loss. The goal of this diet is to make the ketone bodies as an alternative source of energy for the brain instead of glucose. Ketone bodies generated penetrate the blood-brain barrier and provide an alternative source of energy for brain metabolism.17 The ketogenic diet should be started as early as possible and continued at least until adolescence. Although the treatment with ketogenic diet shows a significant decrease in seizure frequency and noticeable improvement in movement disorders, there are many studies claiming that there is an increase in the potential risk of cardiovascular issues and cholesterol level.18 On the other hand, Spalice and Guido19 prospective study over 10 years showed that initial dyslipidemia would resolve over time, and the thickness of carotid intima-media does not increase during the long term management with a ketogenic diet. However, according to Bekker et al.20 cohort study, the failure of a ketogenic diet is dependent on the advanced age at seizure onset, time of the ketogenic diet therapy initiation, and when the diagnosis of GLUT1 deficiency syndrome is established. In addition, they recommend in their study to consider the termination of therapy once there is a deterioration of the EEG findings with no decrease in seizure frequency.
Limitations to our study include difficulty in screening the whole family for the same genetic variant and lack of performing a functional assessment of the variant in animal models. In addition, para-clinical testing such as FDG-PET was not performed due to patient- and hospital-related barriers.
In conclusion, we report a novel heterozygous intronic variant c.1278+12delC in the SLC2A1 gene. This is considered the first report coming from the Middle East and Arab countries. Our study enriches the mutation-spectrum of the SLC2A1 gene and stresses on the importance of whole-exome sequencing in the diagnosis of genetic epilepsies. Early diagnosis and initiation of a ketogenic diet are important goals for the successful management of patients with GLUT1 deficiency syndrome.