Introduction
The 15q11–q13 locus is an area particularly susceptible to rearrangment through non-allelic homologous recombination between low copy repeat sequences leading six structural variation breakpoints (BPs; BP1–BP6), and three further BPs from BP3 to BP5 mapped in this region.1,2 The first description of 15q13.3 microdeleletion (microdel15q13.3) syndrome (OMIM 612001) was reported by Sharp et al.,2 who described nine individuals affected by minor facial features, mild to moderate intellectual disability, epilepsy and/or abnormal electroencephalography (EEG) findings. In this study were examined six individuals which two subjects showed a de novo deletion, two had inherited the molecular anomaly by an affected parent, and in other two the inheritance was unknown.2 Since then, more than a hundred new cases have been reported.3 More recently, a wide study on microdel 15q13.3 was reported by Lowther et al.,4 who collected 246 cases (133 children and 113 adults) plus seven novel adult individuals from local cohort. Developmental delay/intellectual disability (DD/ID) was present in 57.7%, epilepsy/seizure in 28%, speech problems in 15.9%, autism spectrum disorder (ASD) in 10.9%, schizophrenia (SCZ) in 10.2%, mood disorder in 9% and attention deficit hyperactivity disorder (ADHD) in 6.5%.4 In the control group of 23,838 adults no cases of BP4–BP5 deletions were found. Also, in the group of individuals of young age, the study of Lowther et al.,4 underlined the high frequency of behavioral disorders such as ADHD, aggressivity/rage, self-mutilation, poor concentration and poor attention, impulsivity, and other similar features affecting the 34.6% of the cases. Other disturbances included speech problems (13.5%), hypotonia (13.5%), brain anomalies (7.5%), and mood disorders (9.5%). To note no statistically significant differences were observed between the three groups of BP4–BP5, BP3–BP4, and BP4–BP5 deletions.4 The typical 1.6 Mb deletion harbors at least seven genes (ARHGAP11B, MTMR15, Myotubularin-related protein 10 [MTMR10], TRPMI, Kruppel-Like transcription Factor 13a [KLF13], OTUD7A, and cholinergic receptor nicotinic alpha 7 subunit [CHRNA7]), known to contribute to the phenotypic manifestations associated with 15q13.3-deletion syndrome.5,6 Among them, the CHRNA7 gene is deemed the major candidate gene responsible for the presentation of clinical features in individuals with microdel 15q13.3 syndrome.7
According to the work of van Bon et al.,6 15q13 microdeletion were found in 0.02% in normal controls and approximately of the 0.3% in subjects with ID. In seven of 10 siblings from four different families were observed: one showed a mild development delay, four had only learning problems, two without learning problems and only two out of 17 probands had seizures problems. In general, the clinical feature of the microdel 15q13.3 is wide enough ranging from healthy to severe affected individuals with DD/ID, epileptic seizures, ASD, SCZ, behavioral problems and speech dysfunction as the prominent disturbances. Relevant congenital malformations are uncommonly reported.5,8–11
Recently, we have described a four-member family plus an additional unrelated boy with 15q BP3 to BP5 deletions (arr [hg19] 15q13.1–q13.3 [29,213,402–32,510,863]x1).3 The subjects examined have reported a wide range of clinical features such as mild-moderate ID, behavioral disturbances, speech delay and language impairment. In that report, the language dysfunction was highlithed as a notable feature of the 15q13.3 syndrome as also suggested by the observation reported by Pettigrew et al.12
We hereby present a 9-year-old girl followed-up for 9 years affected by microdel 15q13.3, who displayed epileptic seizures of nocturnal frontal lobe epilepsy (NFLE) type, migraine with aura, in association with minor facial features, mild cognitive and language impairment, and circumscribed areas of hypertrichosis. After 9-years follow-up, no more epileptic seizures were reported. A mild improvement was reached in language impairment and hypertrichosis. Still, the girl continues to complain about episodes of migraine. A literature review of the cases involving the region BP4–BP5 is reported.
Case Report
Ethical approval
The study was conducted ethically in accordance with the World Medical Association Declaration of Helsinki and was approved by the ethic committee of the University of Catania, Italy (Ethical Committee Catania 1 Clinical Registration n. 95/2018/PO). Informed consent was obtained from parents of the proband.
Case presentation
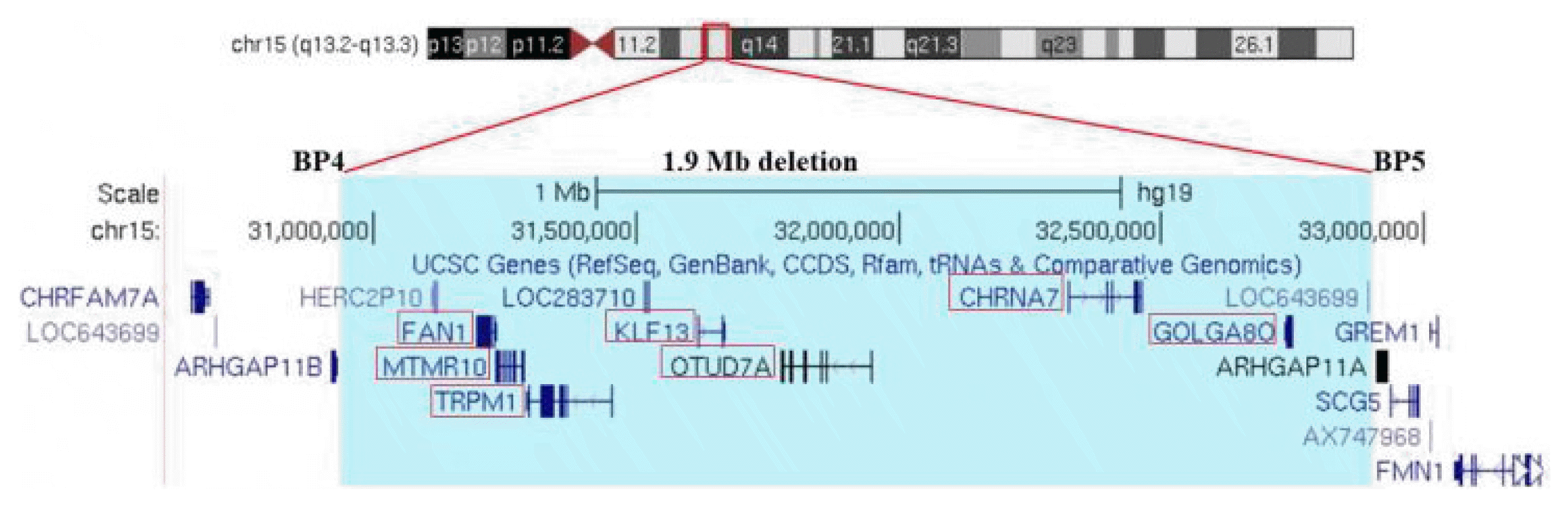
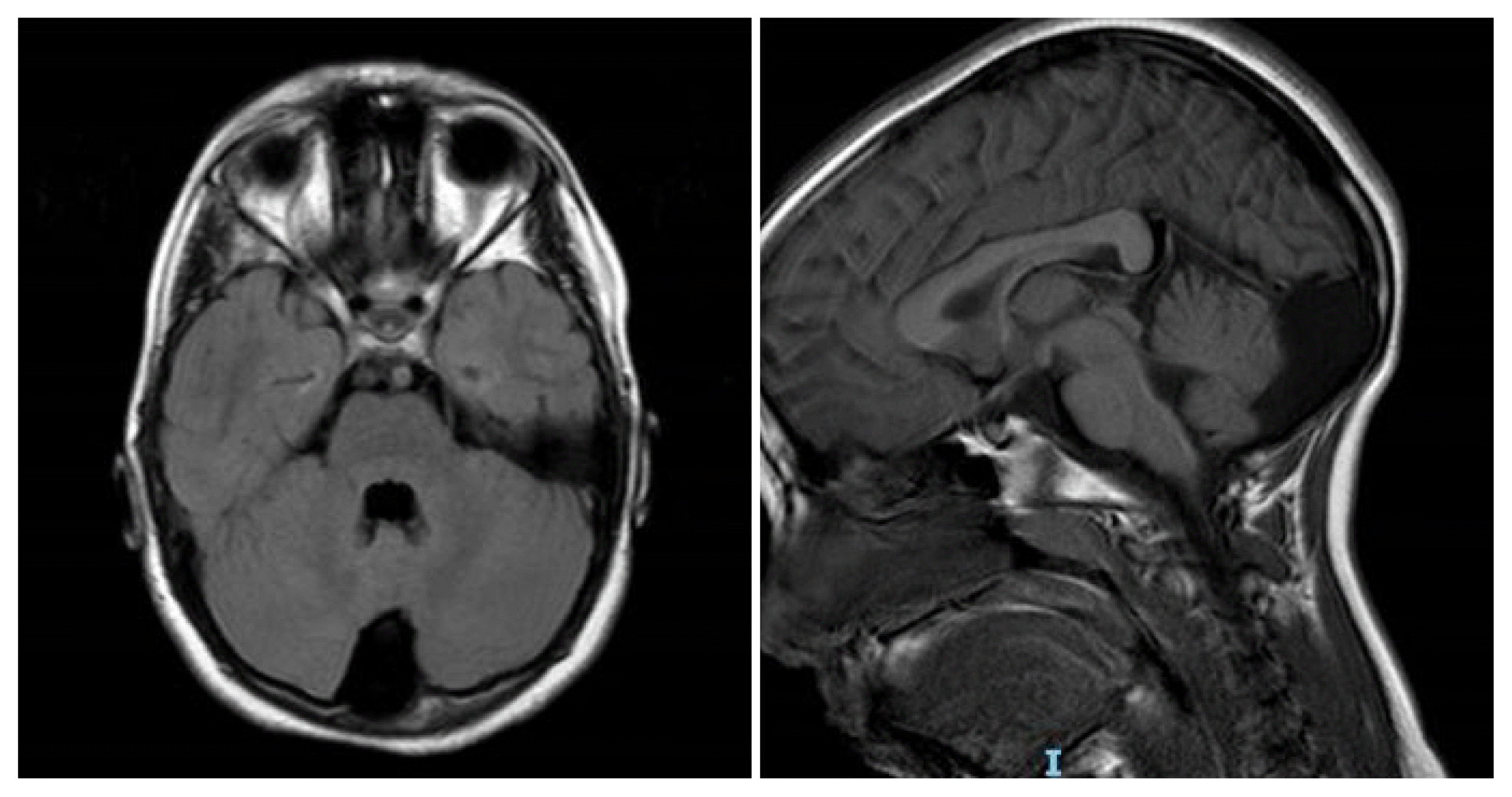
The 9-year-old girl is the third born of healthy unrelated Italian parents. The older sister and brother are healthy. During her pregnancy, the mother referred of being suffered from major depression episodes during the gestation, but having refused any medication help. Neither gestosis, infections, alcohol intake or substance abuse were reported. Normal fetal movements were felt. The girl was born at the 38th weeks of gestation by scheduled cesarean section. Her birthweight was 3,600 g (50th percentile), height was 50 cm (75th percentile), with an occipital frontal circumference (OFC) of 35 cm (50th percentile). The Apgar score was 8 and 10 at 1 and 5 minutes, respectively. The developmental milestones were slightly delayed: first word at 18th month and walking without support at 22th month. At 3 years because of her speech delay, she was referred to a speech therapist rehabilitation with fairly good results. At 9 years, she was admitted to the Pediatric Unit of the Hospital “Policlinico-Vittorio Emanuele” Catania (Italy) for a clinical check-up due to her developmental delay, speech disorder and a poor scholastic performance. Moreover, her parents referred that in the last 2 months their daughter was suffering from several episodes of headache preceded by scotomas lasting several hours with a frequency of once a week and they have also noticed nocturnal episodes of abnormal movements with abrupt awakening. At physical examination the girl was in good conditions. Her weight was 45 kg (<90th percentile), height was 140 cm (90th percentile), and OFC of 52 cm (50th percentile). Minor facial features consisted of narrow forehead size with protruding metopic structure. The teeth were crowded and dyschromic with small eyeteeth. The eyelids were thick particularly in the median region. Abundant panniculus adipose body was seen mainly in the abdomen and in the breast area. Circumscribed areas of hypertrichosis with presence of thick vellus hair in the extensor surfaces of both upper extremities were noted, covering all her shoulder, the forehead, and the supralabial area. Left convex cervico-thoracic scoliosis, bended shoulder and shorter spinal cervico-thorax level was observed. Hands and feet were small. Neurological examination revealed mild hypotonia with coarse walking, normally active patellar reflexes, mild cognitive delay and speech difficulty. All routine laboratory findings including blood count, electrolytes, plasma and urinary amino acid levels, thyroid testing, organic acids, plasma purines, and total cholesterol were normal. No anomalies were found at the electrocardiogram and cardiac ultrasonography. Genital organs were normal and liver and spleen were in normal limits. In the girl the array comparative genomic hybridization (aCGH) analysis dislayed a de novo deletion in the critical deleted region BP4–BP5 at 15q13.2–13.3 (chr 5:30,938,215–32,914,081, UCSC Feb. 2009 hg 19 build) (Fig. 1). Electromyography, nerve conduction velocity test and ophthalmologic examination were normal. EEG recording at the wakefulness (Fig. 2A) and during sleep (Fig. 2B) displayed spike and wave discharges in the fronto-central right region. During the sleep, the abrupt onset of stereotyped hypermotor movements of brief duration involving arms and legs was recorded, associated to vocalization and emotional facial expression with abrupt resolution. Three hours later a new episode was registered. Once awake the girl appeared uncorfortable and irritable after the seizure. The girl and her parents were molecularly karyotyped. Brain magnetic resonance imaging (MRI) revealed right side wide cisterna magna with right retrocerebellar arachnoid cyst (Fig. 3). Neuropsychiatric evaluation showed IQ of 76 on the Wechsler Intelligence Scale for Children (WISC-III). Language was poorly expressive with scanty sentences and insufficient word structures. The girl showed unwillingness to talk to unknown people. Treatment with topiramate was started as the usual recommanded dosage (25–50 mg/day) showing poor response. Due to the lack of efficacy, the use of topimirate was withdrawn and gradually replaced by carbamazepine, application of which decreased both headache attacks and seizures with a normalization of the EEG pattern. We then continued following up the girl (last performed at 18 years of age). The anticonvulsant treatment was gradually suspended. Now at 18 years, her EEG record is normal and no seizures are registered. There is also a mild improvement in language and a spontaneous partial regression of vellus hair. She complains till now of episodes of headache with aura.
Microarray experiment and data analysis
Genomic DNA was isolated from peripheral blood of the proband and parents to analyze structural anomalies using high-resolution CytoSure ISCA 8×60 K microarray from Oxford Gene Technology (OGT). aCGH was performed according to the manufacturer’s recommendations (Agilent Technologies, Santa Clara, CA, USA). Data analysis was done using CytoSure software (GRCh38 assembly; Oxford Gene Technology, Oxford, UK) provided by OGT.
For the copy number variants interpretation were used publicly available patient data on Database of Genomic Variants (dgv.tcag.ca), DECIPHER web-based resource (decipher.sanger.ac.uk), CNV dataset from Clinical Genome Resource and Morbidity Map of Developmental Delay.
The aCGH revealed in the proband a 1.9 Mb deletion of chr15q13.2–13.3(chr15:30,938,215–32,914,081, hg19) harboring seven genes (Fanconi-associated nuclease 1 [FAN1], MTMR10, Transient receptor potential cation channel subfamily M member 1 [TRPM1], KLF13, OTUD7A, CHRNA7, and Golgin subfamily A member O [GOLGA8O]) (Fig. 1). The aCGH analysis of DNA of parents yielded normal results confirming that the proband’s rearrangement was de novo.
Data collection process
According to preferred reporting items for systematic reviews and meta-analyses (PRISMA) statement, 12 publications of reported cases in which BP4–BP5 deletion were identified more of two affected subjects were retrieved through PubMed, Cochrane Library, Scopus, and Web of Science database. Main clinical features collected from literature survey were summarized in Table 1 including the present study.
Discussion
The girl presented a complex series of disturbances consisting of mild ID, marked delayed speech, minor facial features, circumscribed hypertrichosis, migraine with aura and epileptic seizures of NFLE type. In this girl, the clinical course of the epileptic seizures was favourable with good response to antiepileptic treatment, although at a recent assessment, the girl still complains of episodes of migraine with aura. aCGH analysis revealed in the girl a de novo deletion at 15q13.2–13.3 (chr.15: 30,938,215–32,914,081; hg19) with some of critical genes (FAN1, MTMR10, TRPM1, KLF13, OTUD7A, CHRNA7, and GOLGA8O) involved in many neurological features associated to the microdel15q13.3 syndrome (Fig. 1).8,13 Aside from the leading candidate of CHRNA7 gene in neurodevelopmental, neuropsychiatric and epilepsy disorders,1,6,14 it was interesting to find new potential connections of the genomic content to the clinical phenotype showed by the proband. Analyzing potential genotype-phenotype correlations, the role of DNA repair enzyme coded by FAN1 gene prevents from the chromosomal fragility and pathological effects of chromosomal breakpoints. FAN1 deficiency causes DNA damage and abnormalities in repair processes of double-stranded DNA breaks, thus increasing the susceptibility to neurological impairments, such as SCZ or ASD.15 MTMR10 gene belongs to the protein tyrosine phosphatases family, and although being catalytically inactive, it may be somehow important in the neuronal brain dopaminergic neuronal pathways and the phosphorylation activity of dopamine receptors as identified in pathoetiologic mechanisms of SCZ.16,17 There is a clinical relevance in epileptogenesis for the genes TRPM1 and KLF13. A functional consequence of the loss of TRPM1 and KLF13 genes are related to the cortical visual impairment,4,18 and cardiac abnormalities6 observed in some microdel 15q13.3 carriers, respectively. Transient receptor potential channels are expressed both in the nerve terminals of peptidergic nociceptors, and in keratinocytes of epidermis and hair apparatus, suggesting a putative role of TRPM1 gene in the migraine mechanism,19 and in hypertrichosis,20 respectively. The encoded protein by GOLGA8O has been predicted to be involved in Golgi organization and spindle assembly. Accumulating evidences have established the functional involvement of golgins in neuronal excitability, polarity, migration and stress response addressing their contribution to neurological diseases.21–23
There is a notable variety of clinical manifestations of the microdel 15q13.3 and comprises a normal phenotype, borderline IQ, severe cognitive impairment, autism, epileptic seizures, bipolar disorders and schizophrenia.24 The 15q13.3 heterozygous microdeletion syndrome includes a complex and heterogeneous pattern of clinical manifestations with a range of neurodevelopmental disorders, among which cognitive deficits are the most prevalent ones. Epilepsy and behavioral disorders are also frequently reported.14 The variable phenotypic expressivity and degree of severity is related to incomplete penetrance.25 Individuals carrying homozygous microdel 15q13.3 show to have more severe phenotypes in terms of cognitive and behavioral decline, motor dysfunction, cortical visual impairment, and epilepsy.3 In order to provide a deeper examination of abnormal phenotypes associated with the BP4–BP5 rearrangement, we systematically reviewed clinical findings showed by individuals previously reported and included the data of the present girl.
Data collected by the literature are summarized in Table 1.1,2,4,6,9–11,14,24,26–28 Case-reports with less than two subjects examined are not included. Among 117 individuals, the most frequent clinical manifestations were represented by DD/ID (76.9%), behavioral abnormalities (51.2%), speech impairment (42.7%), and epilepsy (30.7%). Facial features (13.6%), ocular impairment (5.9%), and hearth involvement (3.4%) are less frequently reported. Severe congenital malformations are uncommonly found. The data obtained by these reports on clinical features related to 15q13.3 deletions are variable and certainly influenced by selective system of the casuistry carried out. As mentioned above, epileptic disorders are one of the most present clinical features in individuals with microdel 15q13.3, but to our knowledge, only a single case of NFLE related to this disorder has been described so far.27 In a screening of 1,040 individuals with DD/ID of unknown etiology, Sharp et al.2 identified nine individuals with microdel 15q13.3. The evaluation of the clinical spectrum was quite homogeneous and consisted of mild/moderate DD/ID in 9/9 patients, and epilepsy in 6/9. Among 12 patients with epileptic seizures reported by Helbig et al.,10 four were affected by myoclonus plus generalized tonic clonic seizure (GTCS); three by absence; three by absence plus GTCS; and two by absence plus myoclonic seizure and GTCS. Dibbens et al.11 described seven cases: three with juvenile myoclonic epilepsy; three with childhood absence epilepsy, and one with juvenile absence epilepsy. A single case of epilepsy was reported by Ben-Shachar et al.;14 two cases by van Bon et al.,6 and another one by Hassfurther et al.28 In a group of 18 individuals with microdel 15q13.3, Ziats et al.27 reported three cases with epileptic seizures: one with absence, one with GTCS, and one with NFLE. In most of these cases, epileptic treatment was followed with the standard practice according to the age of the affected individuals, the type of epilepsy and its related oucome.29 Brain MRI abnormalities are uncommonly reported in case of BP4–BP5 deletion. In the study of van Bon et al.,6 brain MRI was required in six out 15 individuals with the microdeletion and only three presented abnormalities: arachnoid cyst, parenchymal hypoplasia, subcerebellar picking with dislocation of parenchymal structures, lateral ventral dilatation, and mild corpus callosum agenesis; diffuse changes periventricular and subcortical white matter in the left parietal and temporal region; unspecific increase size of the subarachnoideal space. In the proband, the brain MRI disclosed a right side cisterna magna with right cerebellar arachnoid cyst, findings which seem not to be correlated with the phenotype of epilepsy showed by the girl.
In this study, the girl presented a clinical diagnosis of NFLE, also named sleep-related hypermotor epilepsy. NFLE is a focal epilepsy mainly manifestating in childhood age with clinical episodes, which have abrupt occurence and a brief duration (usually about few seconds) and are typically sleep-related during non-rapid eye movement. The seizures show with motor complex pattern involving arms and legs with ambulatory movements associated to whispering, vocalization or screaming. In some cases, seizures may occur with asymmetric tonic, dystonic or tonic-dystonic posture. These episodes may be recurrent during the same night and may appear during the awakeness.30 Differential diagnosis with sleep disorders is not easy and needs during the episodes a direct witness of expert in this field.31 A great help is given by the EEG and video EEG. The crises arise from the frontal region, but may also have temporal, operculoinsular and posterior origin.29
NFLE may be sporadically expressed or be inherited by autosomal dominant transmission (ADNFLE). Kurahashi and Hirose30 suggested the treatment of NFLE with carbamazepine as they reported a remission of the seizures in about 70% of cases. In the proband we used carbamazepine following the advice of these authors30 obtaining good results. Mutations in CHRNA4 gene coding for the alfa-4 subunit of the neuronal nicotinic acetylcholine receptor have been widely shown to cause ADNFLE.31 In this disorder, recent evidences have also identified a number of heterozygous variants in CHRNB2, CHRNA2, KCNT1, DEPDC5, and CRH genes.32,33 The proband with microdel 15q13.3 complained of NFLE seizures type as one of the three cases reported by Ziats et al.27 The proband showed others peculiar symptoms, which may be likely related to the microdel 15q13.3: the language impairment and hypertrichosis. As reported in Table 1, the language impairment in subjects with BP4–BP5 region was identified in 42.7% of cases and this may be mostly correlated to cognitive impairment presented by these subjects. However, in the proband, the language impairment of the girl was significantly more relevant than her cognitive delay. In reporting the features of four family members and unrelated boy presenting BP3–BP5 deletion, it was noted that the language delay was more pronounced compared to the level of intellectual disability suggesting a likely genotype-phenotype correlation.3
This hypothesis has been confirmed by a report of Pettigrew et al.,12 according to which in a screening cohort of 85 children with language disorder one individual with microdel 15q13.3 was identified presenting language delay in absence of ID. As regards to the congenital hypertrichosis observed in the girl, a similar finding reported by Sharp et al.2 was seen in one out nine cases. Hypertrichosis is defined as an excessive growth of hair of the type terminal, vellus or lanugo in areas of the body that are not predominantly androgen dependent.34 Hypertrichosis has been distinguished in congenital, late-onset, generalized, localized, inherited or acquired.35,36 In the girl, hypertrichosis was congenital and mainly circumscribed to arms and shoulder and less visible in the face. As no cases of headache has been reported in subjects with microdel 15q13.3 syndrome, this finding observed in the girl should be considered as incidental. The course of the symptoms presented by the girl has been particularly benign as concerns the epileptic seizures, which have shown a progressive improvement under anticonvulsant therapy after nine-years follow-up with a clinical and EEG pattern normalization. Mild improvement has also been noted in the cognitive and language disturbances whereas episodes of migraine remain stable.
The girl with the microdel 15q13.3 showed some peculiar symptoms such as NFLE, ID, language delay, hypertrichosis, which has been reported in individuals with the same microdeletion. There is a reason to hypothesize a likely correlation between genotype and phenotype. Limitations of this study are still represented by the absence of new information on the microdel 15q13.3 syndrome and the role of the rearrangement in causing a such unpredictable and variable phenotype. However, similar observations are worthwhile to confirm or revise the clinical information reported.