Introduction
Seizures are transient changes in neurological status caused by hypersynchronous neurons. Epilepsy is a disorder diagnosed when two or more unexplained seizures occur or one seizure with a high risk of recurrence. Epilepsy is one of the commonest neurological disorders that affect around 1% of the population, and most of them start in early childhood and adolescence. The pathophysiology of the disease is still not well understood.1 Epilepsy in Saudi Arabia was reported to be at 6.5 per 1,000 population, which is considered a little bit high in comparison with developed world countries. Most of the epilepsy cases in Saudi Arabia are idiopathic, most probably related to consanguinity and an increased percentage of young people in the Saudi population.2 The relationship between genetic changes and epilepsy is very complex, but genetics play a very important role in disease understanding. Nowadays, scientists developed multiple methods to study genetic changes in these patients which help us to have a closer view and deeper understanding of these disorders. Hereditary epilepsy is divided into two categories; symptomatic and idiopathic. The first category is symptomatic epilepsy, which includes all epilepsies that occur due to a genetic mutation that induces anatomical or pathological changes leading to epilepsy. The second group is idiopathic epilepsy, which includes epilepsies that occur purely due to genetic changes without significant anatomical or pathological abnormalities.3 In this article, we report a Saudi patient with epilepsy and hearing loss who has a heterozygous variant in the calcium voltage-gated channel subunit alpha 1 H (CACNA1H) gene.
Case Report
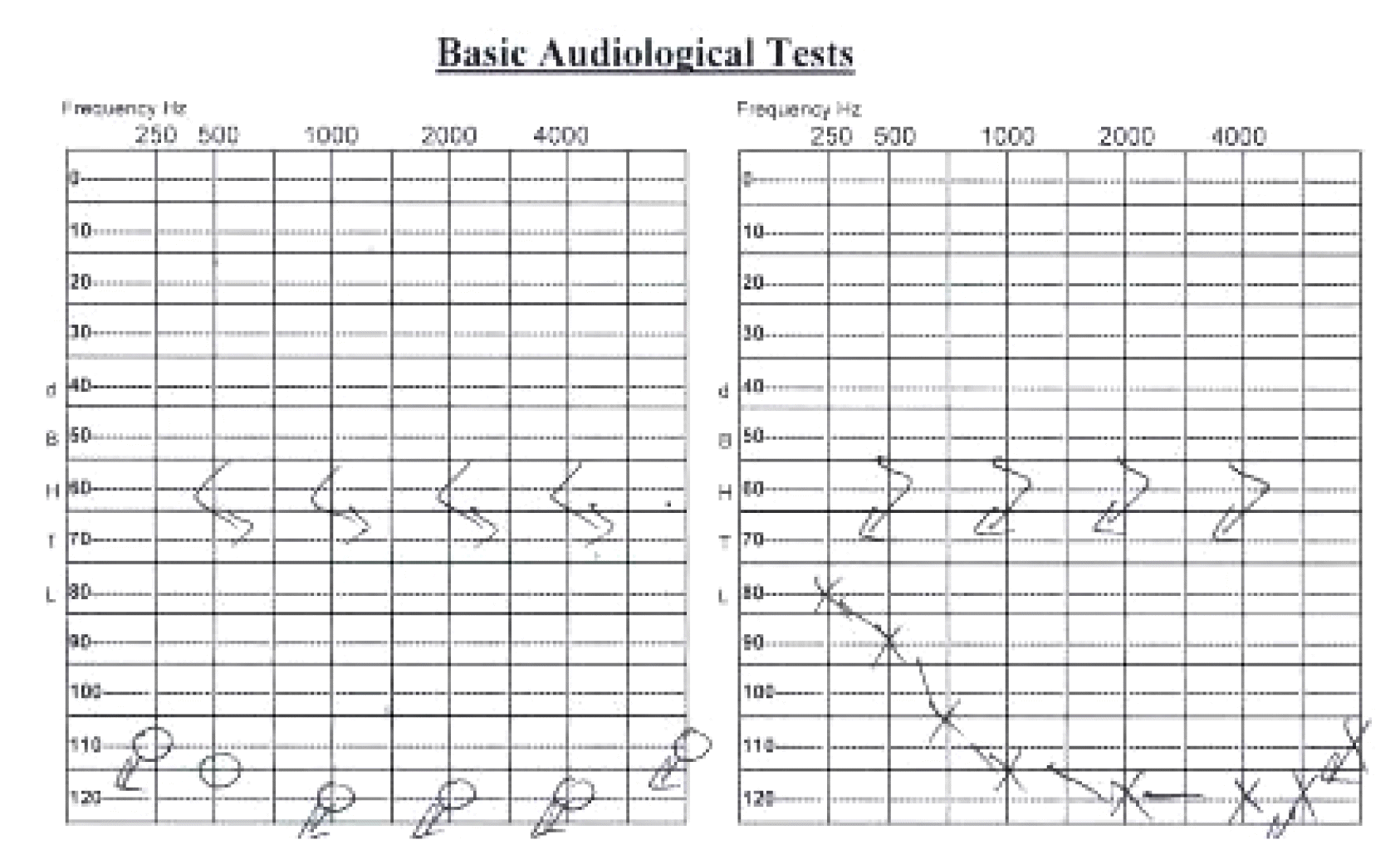
A 50-year-old female presented to the neurology clinic with a history of hearing loss since the age of 4 years and seizures that started 18 years prior. Her seizures are usually induced by emotional stress, prolonged use of the mobile phone, and trauma. She experienced aura before most of the attacks in the form of flashing lights. During attacks, she has loss of consciousness, tonic posturing, clonic movements, and tongue biting with no history of urine or stool incontinence. Each attack lasts for almost 5–10 minutes, and the patient becomes confused with old memory flashbacks and muscle pain for 20–30 minutes after the attacks. The patient reported a frequency of 2–3 seizures per day before starting the medications. She was started on gradually increasing doses of levetiracetam to 500 mg twice daily and carbamazepine 600 mg twice daily. Two months after starting medications, she was seen in the clinic with remarkable improvement of her seizure frequency having only two attacks per month. She had an unremarkable medical and surgical history with no previous history of blood transfusion. There was no family history of a similar condition or chronic neurological disease. She did not smoke and did not have any known allergies. Her general examination showed an obese lady with normal vital signs, integumentary system, and systemic examination including chest, heart, and abdomen with no organomegaly. Her eye and neurological examination were normal apart from bilateral hearing loss. Detailed biochemical and endocrine tests were unremarkable. The hearing assessment showed bilateral profound sensorineural hearing loss (Fig. 1). Computed tomography scan and magnetic resonance imaging of the brain were unremarkable. Molecular genetic analysis of whole exome sequencing identified a heterozygous variant c.395G>A p. (Arg132His) in the CACNA1H gene (OMIM: 607904), which leads to an amino acid exchange. Sixteen out of 22 bioinformatic in silico programs predict a pathogenic effect for this variant. There was no other identified genetic variant associated with hearing loss. Electroencephalogram (EEG) performed twice was normal with no epileptiform discharges or electrographic seizures.
Discussion
Calcium controls a number of essential neuronal responses including neurotransmitter release from the presynaptic terminal and regulation of neuronal excitability. Voltage-gated calcium channels mediate calcium entry into neurons. There are six types of voltage-gated calcium channels.4 The CACNA1H is a gene present in eukaryotic cells located on chromosome 16 that encodes the T-type calcium channels, which are important for calcium influx and depolarization of cells. This gene is spliced at 12–14 sites which generate several different isoforms with distinct properties, and therefore the location of the mutation influences its effects.5
Pathogenic variants in CACNA1H cause autosomal dominant susceptibility to idiopathic generalized epilepsy 6 (OMIM: 611942), which is a broad term that encompasses several common seizure phenotypes, classically including childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy, and epilepsy with grand mal seizures on awakening.6 These recurrent seizures occur in the absence of detectable brain lesions and/or metabolic abnormalities. Seizures are initially generalized with bilateral, synchronous, generalized, and symmetrical EEG discharges.7 Our patient description and semiology are that of focal seizures arising from the occipital lobe with secondary generalization. However, this cannot be confirmed since continuous EEG monitoring in an epilepsy monitoring unit was not performed.
Several previous articles reported a possible association between mutations in different subunits of the voltage-gated calcium channel and epilepsy. However, most evidence for calcium channel involvement in epilepsy has been for CACNA1H.4 Chourasia et al.7 reported the clinical characteristics of five patients with CACNA1H mutations who experienced different types of seizures including absence, focal seizures without awareness, focal seizures with secondary generalization, and myoclonic, atonic, and generalized tonic-clonic seizures. These cases expanded the phenotypic spectrum of CACNA1H as a gene associated with susceptibility to develop generalized epilepsy and focal or multifocal epilepsy of varying severity. Previous studies reported that mutations in CACNA1H affect channel properties directly through altering neuronal electrical properties and indirectly by changing gene expression. Some of the observed functional changes were small with no major biophysical changes, which may indicate that mutations in the CACNA1H gene alone may not be the sole cause of epilepsy but may be considered a risk factor.8,9
The T-type calcium channel is expressed in several organs in the body including kidneys, liver, heart, and brain.10 Nie et al.11 reported the molecular identity and functional properties of the T-type calcium channel cloned from the sensory epithelia of the mouse inner ear. The association between CACNA1H and hearing loss has not been previously reported in the literature. This is the first study that reports a patient with both epilepsy and hearing loss. This indicates that variant in CACNA1H may be the cause of early childhood sensorineural hearing loss in our patient.
Previous studies about patients with epilepsy and CACNA1H mutations reported treatment with a wide variety of antiepileptic medications including ethosuximide, phenobarbital, clobazam, valproic acid, lamotrigine, and oxcarbazepine.7 Some patients still experience seizures despite being on multiple antiepileptic medications. This drug resistance in patients with CACNA1H mutations is multifactorial.12 In our case, we selected the antiepileptic carbamazepine since our patient had focal seizures in which carbamazepine is first-line therapy. Furthermore, we added levetiracetam to achieve better control of her seizures with synergistic effect, different mechanism of action, and limited side effects and drug-drug interactions. The patient’s seizure control significantly improved on this regimen with minimal side effects.
In this article, we reported a Saudi female with a heterozygous variant in the CACNA1H gene (OMIM: 607904) who had epilepsy and hearing loss. This is the first case to report the association of epilepsy and hearing loss with a variant in CACNA1H. We believe that variant may be the reason for developing both epilepsy and sensorineural hearing loss. Further studies are needed to identify the role of CACNA1H in the physiology of the ear to allow for a better understanding of the effects of mutations. In children who present with early childhood hearing loss, genetic studies in highly selected cases including those with a strong family history of hearing loss and epilepsy may help detect a CACNA1H variant early and help with the early screening and diagnosis of other associated disorders including subclinical epilepsy.